
You can not select more than 25 topics
Topics must start with a letter or number, can include dashes ('-') and can be up to 35 characters long.
 linzipeng
4b57f1f031
uodate bed-region
linzipeng
4b57f1f031
uodate bed-region
|
4 年之前 | |
|---|---|---|
| assets | 4 年之前 | |
| tasks | 4 年之前 | |
| README.md | 4 年之前 | |
| inputs | 4 年之前 | |
| workflow.wdl | 4 年之前 | |
README.md
Author : Zipeng Lin
E-mail:18210700121@fudan.edu.cn
Git: http://choppy.3steps.cn/linzipeng/Target-Seq-FUSCC
Last Updates: 09/17/2020
简介
sjzp-target是专用于室间质评项目的,根据 Sentieon 软件靶向测序somatic突变数据分析推荐流程所构建的 Choppy-pipe 系统的 APP。利用该 APP 可以获得从全基因组测序原始文件fastq 到包含somatic突变信息的vcf文件的整个过程。主要包括数据的预处理、中间数据质控以及变异的检测。
快速安装
Requirements
- Python 3
- choppy
- Ali-Cloud
在终端中输入以下命令即可快速安装本APP。
$ source activate choppy-pipe-0.3.8.dev0
$ choppy install linzipeng/Target-Seq-FUSCC
$ choppy apps
使用方法
任务准备
按照上述步骤安装成功之后,可以通过下面简单的命令即可使用APP:
# Generate samples file
$ choppy samples linzipeng/Target-Seq-FUSCC-latest --out samples.csv
sample.csv 包含以下几个需要填写的参数:
tumor_fastq_1,tumor_fastq_2,normal_fastq_1,normal_fastq_1,sample_name,cluster,sample_id,disk_size
# tumor_fastq_1 tumor双端测序数据的R1端在阿里云上的路径信息
# tumor_fastq_2 tumor双端测序数据的R2端在阿里云上的路径信息
# normal_fastq_1 normal双端测序数据的R1端在阿里云上的路径信息
# normal_fastq_2 normal双端测序数据的R2端在阿里云上的路径信息
# sample_name 输出文件名的前缀
# cluster 使用的机器类型,不填则默认使用 OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc
# sample_id 每个样本任务的识别码。注意:同一个samples文件中,不同样本的ID应该不同
# disk_size 任务运行时,集群存储空间设置
机器类型选择可以参照:计算网络增强型实例规格族sn1ne 以及 bcs 类型机器,对于全基因组数据不要使用小于32CPU的机器类型
任务提交
在配置好samples.csv 文件后,使用以下命令可以提交计算任务:
$ choppy batch linzipeng/Target-Seq-FUSCC-latest sample.csv --project-name Your_project_name
提交成功后,即可在工作目录下找到生成的目录名为Your_project_name,里面包含了本次提交任务的所有样本信息。
任务输出
任务成功结束后,便可以在阿里云相应的OSS端生成相应的结果文件。包括数据预处理产生的中间结果文件以及变异检测得到的vcf文件。
流程示意图
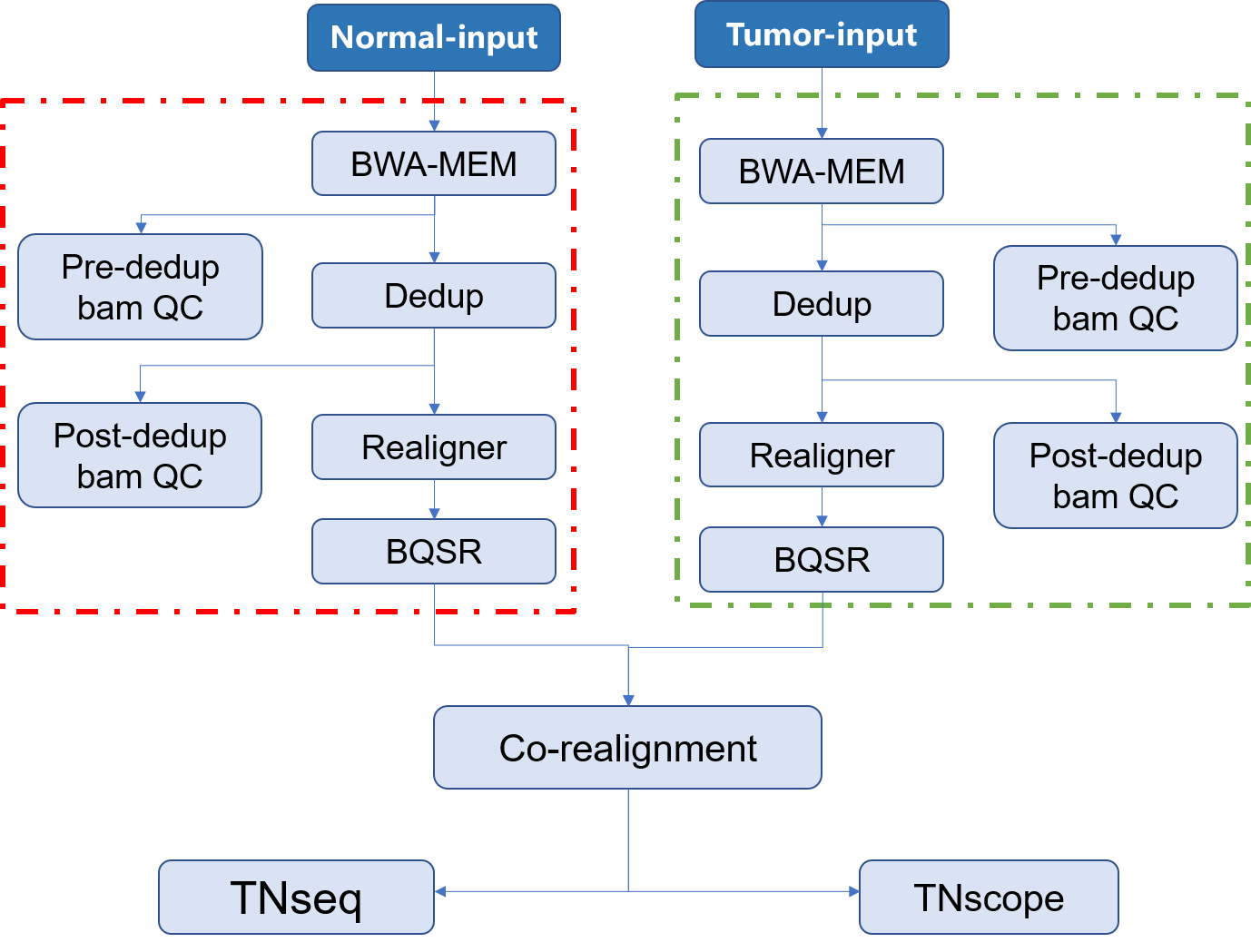
输出文件说明
整个分析流程中,每个步骤输出的结果说明如下:
- call-mapping 原始数据经过比对后生成的排序后的
sample.sorted.bam文件及其索引文件 - call-Metrics 比对后生成的
sample.sorted.bam文件的质控信息 - call-Dedup 比对的结果去除重复后的
sample.sorted.deduped.bam文件及其索引文件 - call-deduped_Metrics 去除重复后的
sample.sorted.deduped.bam文件的质控信息 - call-Realigner 去除重复后重比对的
sample.sorted.deduped.realigned.bam文件及其索引文件 - call-BQSR 局部碱基矫正的
sample.sorted.deduped.realigned.recaled.bam文件、索引文件及其相关信息 - call-TNseq 使用TNseq检测得到的
sample.vcf文件及其索引文件 - call-TNscope 使用TNscope检测得到的
sample.vcf文件及其索引文件
软件版本及参数
| 软件/文件 | 版本 |
|---|---|
| Sentieon | v2019.11.28 |
| 参考基因组(fasta) | hg19 |
| dbsnp | dbsnp_138.hg19_nochr_sorted.vcf |
| db_mills | Mills_and_1000G_gold_standard.indels.hg19_nochr.vcf |