

incheckning
2538bbf78f
15 ändrade filer med 808 tillägg och 0 borttagningar
+ 106
- 0
README.md
Visa fil
| @@ -0,0 +1,106 @@ | |||
| > Author : Zipeng Lin | |||
| > | |||
| > | |||
| > E-mail:[18210700121@fudan.edu.cn](mailto:1721070009@fudan.edu.cn) | |||
| > | |||
| > Git: <http://choppy.3steps.cn/linzipeng/sjzp-target.git> | |||
| > | |||
| > Last Updates: 09/17/2020 | |||
| ## 简介 | |||
| `sjzp-target`是专用于室间质评项目的,根据 [Sentieon](https://support.sentieon.com/manual/) 软件靶向测序`somatic`突变数据分析推荐流程所构建的 [Choppy-pipe](http://choppy.3steps.cn/) 系统的 APP。利用该 APP 可以获得从全基因组测序原始文件`fastq` 到包含`somatic`突变信息的`vcf`文件的整个过程。主要包括数据的预处理、中间数据质控以及变异的检测。 | |||
| ## 快速安装 | |||
| #### Requirements | |||
| - Python 3 | |||
| - [choppy](http://choppy.3steps.cn/) | |||
| - Ali-Cloud | |||
| 在终端中输入以下命令即可快速安装本APP。 | |||
| ```bash | |||
| $ source activate choppy-pipe-0.3.8.dev0 | |||
| $ choppy install linzipeng/sjzp | |||
| $ choppy apps | |||
| ``` | |||
| ## 使用方法 | |||
| ### 任务准备 | |||
| 按照上述步骤安装成功之后,可以通过下面简单的命令即可使用APP: | |||
| ```bash | |||
| # Generate samples file | |||
| $ choppy samples linzipeng/sjzp-target-latest --out samples.csv | |||
| ``` | |||
| `sample.csv` 包含以下几个需要填写的参数: | |||
| ``` bash | |||
| tumor_fastq_1,tumor_fastq_2,normal_fastq_1,normal_fastq_1,sample_name,cluster,sample_id,disk_size | |||
| # tumor_fastq_1 tumor双端测序数据的R1端在阿里云上的路径信息 | |||
| # tumor_fastq_2 tumor双端测序数据的R2端在阿里云上的路径信息 | |||
| # normal_fastq_1 normal双端测序数据的R1端在阿里云上的路径信息 | |||
| # normal_fastq_2 normal双端测序数据的R2端在阿里云上的路径信息 | |||
| # sample_name 输出文件名的前缀 | |||
| # cluster 使用的机器类型,不填则默认使用 OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc | |||
| # sample_id 每个样本任务的识别码。注意:同一个samples文件中,不同样本的ID应该不同 | |||
| # disk_size 任务运行时,集群存储空间设置 | |||
| ``` | |||
| > 机器类型选择可以参照:[计算网络增强型实例规格族sn1ne](https://help.aliyun.com/document_detail/25378.html?spm=a2c4g.11186623.2.11.1a3d6b46rNmksN#sn1ne) 以及 [bcs](https://help.aliyun.com/document_detail/42391.html?spm=5176.10695662.1996646101.searchclickresult.5c0b12b5Adk9Xc) 类型机器,对于全基因组数据不要使用小于32CPU的机器类型 | |||
| ### 任务提交 | |||
| 在配置好`samples.csv` 文件后,使用以下命令可以提交计算任务: | |||
| ```bash | |||
| $ choppy batch linzipeng/sjzp-target-latest sample.csv --project-name Your_project_name | |||
| ``` | |||
| 提交成功后,即可在工作目录下找到生成的目录名为Your_project_name,里面包含了本次提交任务的所有样本信息。 | |||
| ### 任务输出 | |||
| 任务成功结束后,便可以在阿里云相应的OSS端生成相应的结果文件。包括数据预处理产生的中间结果文件以及变异检测得到的`vcf`文件。 | |||
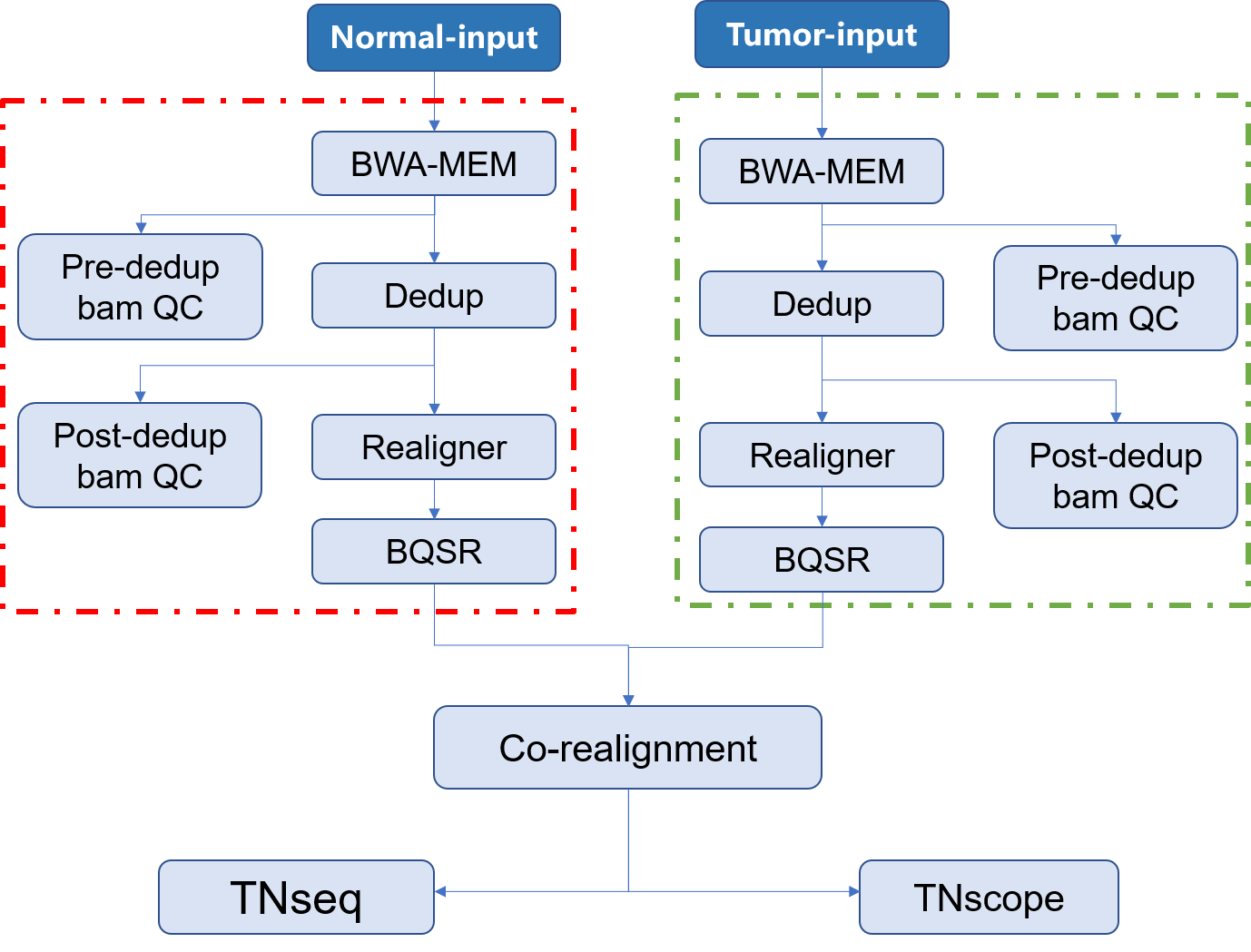
| ## 流程示意图 | |||
|  | |||
| ### 输出文件说明 | |||
| 整个分析流程中,每个步骤输出的结果说明如下: | |||
| - **call-mapping** 原始数据经过比对后生成的排序后的`sample.sorted.bam`文件及其索引文件 | |||
| - **call-Metrics** 比对后生成的`sample.sorted.bam`文件的质控信息 | |||
| - **call-Dedup** 比对的结果去除重复后的`sample.sorted.deduped.bam`文件及其索引文件 | |||
| - **call-deduped_Metrics** 去除重复后的`sample.sorted.deduped.bam` 文件的质控信息 | |||
| - **call-Realigner** 去除重复后重比对的`sample.sorted.deduped.realigned.bam`文件及其索引文件 | |||
| - **call-BQSR** 局部碱基矫正的`sample.sorted.deduped.realigned.recaled.bam`文件、索引文件及其相关信息 | |||
| - **call-TNseq** 使用TNseq检测得到的`sample.vcf`文件及其索引文件 | |||
| - **call-TNscope** 使用TNscope检测得到的`sample.vcf`文件及其索引文件 | |||
| ### 软件版本及参数 | |||
| | 软件/文件 | 版本 | | |||
| | :------------------ | --------------------------------------------------- | | |||
| | Sentieon | v2019.11.28 | | |||
| | 参考基因组(fasta) | hg19 | | |||
| | dbsnp | dbsnp_138.hg19_nochr_sorted.vcf | | |||
| | db_mills | Mills_and_1000G_gold_standard.indels.hg19_nochr.vcf | | |||
| ### 附录 | |||
| ### 参考文献 | |||
Binär
assets/1549784133993.png
Visa fil
{kind=link}
| Before | After |
|---|---|

|
|
| Width: 922 | Height: 464 | Size: 55KB |
Binär
assets/somatic.png
Visa fil
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1378 | Height: 1049 | Size: 97KB |
+ 18
- 0
inputs
Visa fil
| @@ -0,0 +1,18 @@ | |||
| { | |||
| "{{ project_name }}.fasta": "hg19_nochr.fa", | |||
| "{{ project_name }}.ref_dir": "oss://pgx-reference-data/hg19/", | |||
| "{{ project_name }}.dbsnp": "dbsnp_138.hg19_nochr_sorted.vcf", | |||
| "{{ project_name }}.dbsnp_dir": "oss://pgx-reference-data/hg19/", | |||
| "{{ project_name }}.SENTIEON_INSTALL_DIR": "/opt/sentieon-genomics", | |||
| "{{ project_name }}.dbmills_dir": "oss://pgx-reference-data/hg19/", | |||
| "{{ project_name }}.db_mills": "Mills_and_1000G_gold_standard.indels.hg19_nochr.vcf", | |||
| "{{ project_name }}.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/sentieon-genomics:v2019.11.28", | |||
| "{{ project_name }}.sample": "{{ sample_name }}", | |||
| "{{ project_name }}.tumor_fastq_2": "{{ tumor_fastq_2 }}", | |||
| "{{ project_name }}.tumor_fastq_1": "{{ tumor_fastq_1 }}", | |||
| "{{ project_name }}.normal_fastq_1": "{{ normal_fastq_1 }}", | |||
| "{{ project_name }}.normal_fastq_2": "{{ normal_fastq_2 }}", | |||
| "{{ project_name }}.disk_size": "{{ disk_size }}", | |||
| "{{ project_name }}.cluster_config": "OnDemand bcs.a2.3xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.regions": "oss://pgx-source-data/sjzp/2020/141gene_twist_merged.bed" | |||
| } | |||
+ 49
- 0
tasks/BQSR.wdl
Visa fil
| @@ -0,0 +1,49 @@ | |||
| task BQSR { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File regions | |||
| File realigned_bam | |||
| File realigned_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} --interval ${regions} --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} -q ${sample}_recal_data.table --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table.post --algo ReadWriter ${sample}.sorted.deduped.realigned.recaled.bam | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt --algo QualCal --plot --before ${sample}_recal_data.table --after ${sample}_recal_data.table.post ${sample}_recal_data.csv | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot QualCal -o ${sample}_bqsrreport.pdf ${sample}_recal_data.csv | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File recal_table = "${sample}_recal_data.table" | |||
| File recal_post = "${sample}_recal_data.table.post" | |||
| File recaled_bam = "${sample}.sorted.deduped.realigned.recaled.bam" | |||
| File recaled_bam_index = "${sample}.sorted.deduped.realigned.recaled.bam.bai" | |||
| File recal_csv = "${sample}_recal_data.csv" | |||
| File bqsrreport_pdf = "${sample}_bqsrreport.pdf" | |||
| } | |||
| } | |||
+ 40
- 0
tasks/Dedup.wdl
Visa fil
| @@ -0,0 +1,40 @@ | |||
| task Dedup { | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo LocusCollector --fun score_info ${sample}_score.txt | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo Dedup --rmdup --score_info ${sample}_score.txt --metrics ${sample}_dedup_metrics.txt ${sample}.sorted.deduped.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File score = "${sample}_score.txt" | |||
| File dedup_metrics = "${sample}_dedup_metrics.txt" | |||
| File Dedup_bam = "${sample}.sorted.deduped.bam" | |||
| File Dedup_bam_index = "${sample}.sorted.deduped.bam.bai" | |||
| } | |||
| } | |||
+ 38
- 0
tasks/Haplotyper.wdl
Visa fil
| @@ -0,0 +1,38 @@ | |||
| task Haplotyper { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File recaled_bam | |||
| File recaled_bam_index | |||
| String dbsnp | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper -d ${dbsnp_dir}/${dbsnp} ${sample}_hc.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf = "${sample}_hc.vcf" | |||
| File vcf_idx = "${sample}_hc.vcf.idx" | |||
| } | |||
| } | |||
+ 56
- 0
tasks/Metrics.wdl
Visa fil
| @@ -0,0 +1,56 @@ | |||
| task Metrics { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${sorted_bam} --algo MeanQualityByCycle ${sample}_mq_metrics.txt --algo QualDistribution ${sample}_qd_metrics.txt --algo GCBias --summary ${sample}_gc_summary.txt ${sample}_gc_metrics.txt --algo AlignmentStat ${sample}_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_is_metrics.txt --algo CoverageMetrics --omit_base_output ${sample}_coverage_metrics | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot metrics -o ${sample}_metrics_report.pdf gc=${sample}_gc_metrics.txt qd=${sample}_qd_metrics.txt mq=${sample}_mq_metrics.txt isize=${sample}_is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File qd_metrics = "${sample}_qd_metrics.txt" | |||
| File qd_metrics_pdf = "${sample}_qd_metrics.pdf" | |||
| File mq_metrics = "${sample}_mq_metrics.txt" | |||
| File mq_metrics_pdf = "${sample}_mq_metrics.pdf" | |||
| File is_metrics = "${sample}_is_metrics.txt" | |||
| File is_metrics_pdf = "${sample}_is_metrics.pdf" | |||
| File gc_summary = "${sample}_gc_summary.txt" | |||
| File gc_metrics = "${sample}_gc_metrics.txt" | |||
| File gc_metrics_pdf = "${sample}_gc_metrics.pdf" | |||
| File aln_metrics = "${sample}_aln_metrics.txt" | |||
| File coverage_metrics_sample_summary = "${sample}_coverage_metrics.sample_summary" | |||
| File coverage_metrics_sample_statistics = "${sample}_coverage_metrics.sample_statistics" | |||
| File coverage_metrics_sample_interval_statistics = "${sample}_coverage_metrics.sample_interval_statistics" | |||
| File coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File coverage_metrics_sample_cumulative_coverage_counts = "${sample}_coverage_metrics.sample_cumulative_coverage_counts" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/Realigner.wdl
Visa fil
| @@ -0,0 +1,41 @@ | |||
| task Realigner { | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File regions | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String db_mills | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo Realigner -k ${dbmills_dir}/${db_mills} --interval_list ${regions} ${sample}.sorted.deduped.realigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File realigner_bam = "${sample}.sorted.deduped.realigned.bam" | |||
| File realigner_bam_index = "${sample}.sorted.deduped.realigned.bam.bai" | |||
| } | |||
| } | |||
+ 42
- 0
tasks/TNscope.wdl
Visa fil
| @@ -0,0 +1,42 @@ | |||
| task TNscope { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String tumor_name | |||
| String normal_name | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File corealigner_bam | |||
| File corealigner_bam_index | |||
| String dbsnp | |||
| String disk_size | |||
| File regions | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver --interval ${regions} -r ${ref_dir}/${fasta} -t $nt -i ${corealigner_bam} --algo TNscope --tumor_sample ${tumor_name} --normal_sample ${normal_name} --dbsnp ${dbsnp_dir}/${dbsnp} ${sample}.TNscope.TN.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File TNscope_vcf= "${sample}.TNscope.TN.vcf" | |||
| File TNscope_vcf_index = "${sample}.TNscope.TN.vcf.idx" | |||
| } | |||
| } | |||
+ 43
- 0
tasks/TNseq.wdl
Visa fil
| @@ -0,0 +1,43 @@ | |||
| task TNseq { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String tumor_name | |||
| String normal_name | |||
| String docker | |||
| String cluster_config | |||
| File regions | |||
| String fasta | |||
| File corealigner_bam | |||
| File corealigner_bam_index | |||
| String dbsnp | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver --interval ${regions} -r ${ref_dir}/${fasta} -t $nt -i ${corealigner_bam} --algo TNhaplotyper --tumor_sample ${tumor_name} --normal_sample ${normal_name} --dbsnp ${dbsnp_dir}/${dbsnp} ${sample}.TNseq.TN.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File TNseq_vcf= "${sample}.TNseq.TN.vcf" | |||
| File TNseq_vcf_index = "${sample}.TNseq.TN.vcf.idx" | |||
| } | |||
| } | |||
+ 44
- 0
tasks/corealigner.wdl
Visa fil
| @@ -0,0 +1,44 @@ | |||
| task corealigner { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File tumor_recaled_bam | |||
| File tumor_recaled_bam_index | |||
| File normal_recaled_bam | |||
| File normal_recaled_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${tumor_recaled_bam} -i ${normal_recaled_bam} --algo Realigner -k ${dbmills_dir}/${db_mills} -k ${dbsnp_dir}/${dbsnp} ${sample}_corealigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File corealigner_bam = "${sample}_corealigned.bam" | |||
| File corealigner_bam_index = "${sample}_corealigned.bam.bai" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/deduped_Metrics.wdl
Visa fil
| @@ -0,0 +1,37 @@ | |||
| task deduped_Metrics { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File deduped_coverage_metrics_sample_summary = "${sample}_deduped_coverage_metrics.sample_summary" | |||
| File deduped_coverage_metrics_sample_statistics = "${sample}_deduped_coverage_metrics.sample_statistics" | |||
| File deduped_coverage_metrics_sample_interval_statistics = "${sample}_deduped_coverage_metrics.sample_interval_statistics" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_counts = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_counts" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/mapping.wdl
Visa fil
| @@ -0,0 +1,34 @@ | |||
| task mapping { | |||
| File ref_dir | |||
| String fasta | |||
| File fastq_1 | |||
| File fastq_2 | |||
| String SENTIEON_INSTALL_DIR | |||
| String group | |||
| String sample | |||
| String pl | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i - | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File sorted_bam = "${sample}.sorted.bam" | |||
| File sorted_bam_index = "${sample}.sorted.bam.bai" | |||
| } | |||
| } | |||
+ 260
- 0
workflow.wdl
Visa fil
| @@ -0,0 +1,260 @@ | |||
| import "./tasks/mapping.wdl" as mapping | |||
| import "./tasks/Metrics.wdl" as Metrics | |||
| import "./tasks/Dedup.wdl" as Dedup | |||
| import "./tasks/deduped_Metrics.wdl" as deduped_Metrics | |||
| import "./tasks/Realigner.wdl" as Realigner | |||
| import "./tasks/BQSR.wdl" as BQSR | |||
| import "./tasks/corealigner.wdl" as corealigner | |||
| import "./tasks/TNseq.wdl" as TNseq | |||
| import "./tasks/TNscope.wdl" as TNscope | |||
| workflow {{ project_name }} { | |||
| File tumor_fastq_1 | |||
| File tumor_fastq_2 | |||
| File normal_fastq_1 | |||
| File normal_fastq_2 | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String docker | |||
| String fasta | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String db_mills | |||
| File dbsnp_dir | |||
| String dbsnp | |||
| String disk_size | |||
| String cluster_config | |||
| File regions | |||
| call mapping.mapping as tumor_mapping { | |||
| input: | |||
| group=sample + "tumor", | |||
| sample=sample + "tumor", | |||
| fastq_1=tumor_fastq_1, | |||
| fastq_2=tumor_fastq_2, | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| pl="ILLUMINAL", | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Metrics.Metrics as tumor_Metrics { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| sorted_bam=tumor_mapping.sorted_bam, | |||
| sorted_bam_index=tumor_mapping.sorted_bam_index, | |||
| sample=sample + "tumor", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Dedup.Dedup as tumor_Dedup { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=tumor_mapping.sorted_bam, | |||
| sorted_bam_index=tumor_mapping.sorted_bam_index, | |||
| sample=sample + "tumor", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as tumor_deduped_Metrics { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=tumor_Dedup.Dedup_bam, | |||
| Dedup_bam_index=tumor_Dedup.Dedup_bam_index, | |||
| sample=sample + "tumor", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Realigner.Realigner as tumor_Realigner { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| regions=regions, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=tumor_Dedup.Dedup_bam, | |||
| Dedup_bam_index=tumor_Dedup.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| sample=sample + "tumor", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call BQSR.BQSR as tumor_BQSR { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| regions=regions, | |||
| realigned_bam=tumor_Realigner.realigner_bam, | |||
| realigned_bam_index=tumor_Realigner.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| sample=sample + "tumor", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call mapping.mapping as normal_mapping { | |||
| input: | |||
| group=sample + "normal", | |||
| sample=sample + "normal", | |||
| fastq_1=normal_fastq_1, | |||
| fastq_2=normal_fastq_2, | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| pl="ILLUMINAL", | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Metrics.Metrics as normal_Metrics { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| sorted_bam=normal_mapping.sorted_bam, | |||
| sorted_bam_index=normal_mapping.sorted_bam_index, | |||
| sample=sample + "normal", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Dedup.Dedup as normal_Dedup { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=normal_mapping.sorted_bam, | |||
| sorted_bam_index=normal_mapping.sorted_bam_index, | |||
| sample=sample + "normal", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as normal_deduped_Metrics { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=normal_Dedup.Dedup_bam, | |||
| Dedup_bam_index=normal_Dedup.Dedup_bam_index, | |||
| sample=sample + "normal", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call Realigner.Realigner as normal_Realigner { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| regions=regions, | |||
| Dedup_bam=normal_Dedup.Dedup_bam, | |||
| Dedup_bam_index=normal_Dedup.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| sample=sample + "normal", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call BQSR.BQSR as normal_BQSR { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| regions=regions, | |||
| realigned_bam=normal_Realigner.realigner_bam, | |||
| realigned_bam_index=normal_Realigner.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| sample=sample + "normal", | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call corealigner.corealigner as corealigner { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| sample=sample, | |||
| docker=docker, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| disk_size=disk_size, | |||
| tumor_recaled_bam=tumor_BQSR.recaled_bam, | |||
| tumor_recaled_bam_index=tumor_BQSR.recaled_bam_index, | |||
| normal_recaled_bam=normal_BQSR.recaled_bam, | |||
| normal_recaled_bam_index=normal_BQSR.recaled_bam_index, | |||
| cluster_config=cluster_config | |||
| } | |||
| call TNseq.TNseq as TNseq { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| regions=regions, | |||
| corealigner_bam=corealigner.corealigner_bam, | |||
| corealigner_bam_index=corealigner.corealigner_bam_index, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| tumor_name=sample + "tumor", | |||
| normal_name=sample + "normal", | |||
| docker=docker, | |||
| sample=sample, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| call TNscope.TNscope as TNscope { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| regions=regions, | |||
| corealigner_bam=corealigner.corealigner_bam, | |||
| corealigner_bam_index=corealigner.corealigner_bam_index, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| tumor_name=sample + "tumor", | |||
| normal_name=sample + "normal", | |||
| docker=docker, | |||
| sample=sample, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| } | |||
Laddar…