

коміт
af97065da6
9 змінених файлів з 176 додано та 0 видалено
+ 21
- 0
README.md
Переглянути файл
| @@ -0,0 +1,21 @@ | |||
| # CNVkit | |||
| > Author: Yaqing Liu | |||
| > | |||
| > E-mail:yaqing.liu@outlook.com | |||
| > | |||
| ## 安装指南 | |||
| ``` | |||
| # 激活choppy环境 | |||
| open-choppy-env | |||
| # 安装app | |||
| choppy install YaqingLiu/CNVkit | |||
| ``` | |||
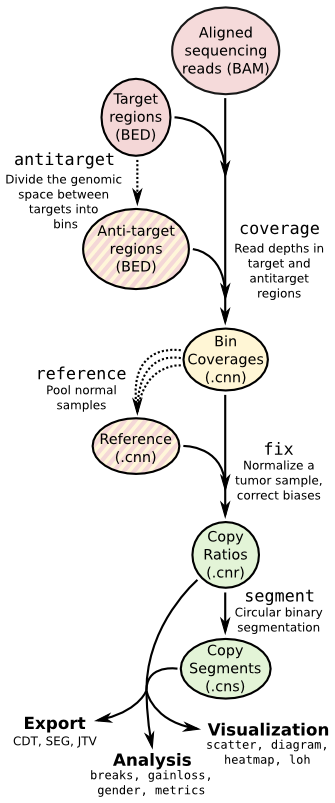
| ## Copy number calling pipeline | |||
| 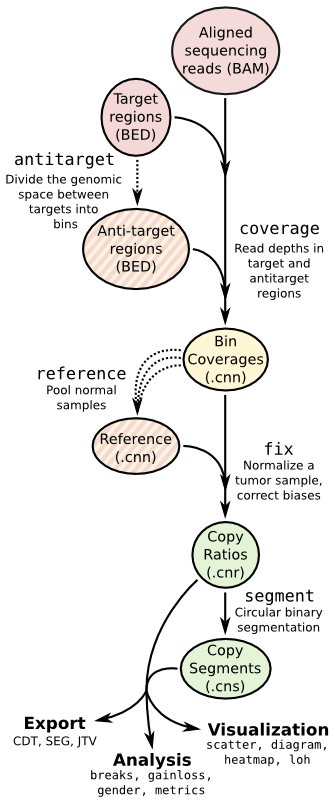 | |||
| ## App输入文件 | |||
BIN
assest/cnv_calling_pipeline.png
Переглянути файл
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 335 | Height: 803 | Size: 82KB |
+ 8
- 0
defaults
Переглянути файл
| @@ -0,0 +1,8 @@ | |||
| { | |||
| "fasta": "GRCh38.d1.vd1.fa", | |||
| "ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "docker": "registry.cn-hangzhou.aliyuncs.com/pgx-docker-registry/cnvkit:0.9.7", | |||
| "disk_size": "200", | |||
| "cluster_config": "OnDemand bcs.a2.3xlarge img-ubuntu-vpc", | |||
| "bed": "oss://pgx-reference-data/reference/wes_bedfiles/agilent_v6/SureSelect_Human_All_Exon_V6_r2.bed" | |||
| } | |||
+ 13
- 0
inputs
Переглянути файл
| @@ -0,0 +1,13 @@ | |||
| { | |||
| "{{ project_name }}.sample_id": "{{ sample_id }}", | |||
| "{{ project_name }}.normal_bam": "{{ normal_bam }}", | |||
| "{{ project_name }}.normal_bai": "{{ normal_bai }}", | |||
| "{{ project_name }}.tumor_bam": "{{ tumor_bam }}", | |||
| "{{ project_name }}.tumor_bai": "{{ tumor_bai }}", | |||
| "{{ project_name }}.fasta": "{{ fasta }}", | |||
| "{{ project_name }}.ref_dir": "{{ ref_dir }}", | |||
| "{{ project_name }}.docker": "{{ docker }}", | |||
| "{{ project_name }}.bed": "{{ bed }}", | |||
| "{{ project_name }}.disk_size": "{{ disk_size }}", | |||
| "{{ project_name }}.cluster_config": "{{ cluster_config }}" | |||
| } | |||
BIN
tasks/.DS_Store
Переглянути файл
+ 22
- 0
tasks/access.wdl
Переглянути файл
| @@ -0,0 +1,22 @@ | |||
| task access { | |||
| File ref_dir | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cnvkit.py access ${ref_dir}/${fasta} -o ./data/access-5kb-mappable.hg38.bed | |||
| >>> | |||
| runtime { | |||
| docker: docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File access_bed = "access-5kb-mappable.hg38.bed" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/batch.wdl
Переглянути файл
| @@ -0,0 +1,37 @@ | |||
| task batch { | |||
| String sample_id | |||
| File tumor_bam | |||
| File tumor_bai | |||
| File normal_bam | |||
| File normal_bai | |||
| String bed | |||
| File ref_dir | |||
| String fasta | |||
| File access_bed | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cnvkit.py batch ${tumor_bam} --normal ${normal_bam} \ | |||
| --targets ${bed} \ | |||
| --fasta ${ref_dir}/${fasta} --access ${access_bed} \ | |||
| --output-reference my_reference.cnn --output-dir ./results/ \ | |||
| --diagram --scatter | |||
| >>> | |||
| runtime { | |||
| docker: docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File reference_cnn = "my_reference.cnn" | |||
| Array[File] result = glob("result/*") | |||
| } | |||
| } | |||
+ 25
- 0
tasks/export.wdl
Переглянути файл
| @@ -0,0 +1,25 @@ | |||
| task export{ | |||
| Array[File] result | |||
| String docker | |||
| String cluster | |||
| String disk_size | |||
| command <<< | |||
| mkdir -p /cromwell_root/tmp/cnvkit_export | |||
| cp -r ${sep=" " result} /cromwell_root/tmp/cnvkit_export | |||
| cd /cromwell_root/tmp/cnvkit_export | |||
| # this version app just provides seg | |||
| cnvkit.py export seg ./results/*.cns -o samples.seg | |||
| >>> | |||
| runtime { | |||
| docker: docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File seg = "samples.seg" | |||
| } | |||
| } | |||
+ 50
- 0
workflow.wdl
Переглянути файл
| @@ -0,0 +1,50 @@ | |||
| import "./tasks/access.wdl" as access | |||
| import "./tasks/batch.wdl" as batch | |||
| import "./tasks/export.wdl" as export | |||
| workflow {{ project_name }} { | |||
| String sample_id | |||
| File tumor_bam | |||
| File tumor_bai | |||
| File normal_bam | |||
| File normal_bai | |||
| String bed | |||
| File ref_dir | |||
| String fasta | |||
| String docker | |||
| String cluster | |||
| String disk_size | |||
| call access.access as access { | |||
| input: | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| docker = docker, | |||
| cluster = cluster, | |||
| disk_size = disk_size | |||
| } | |||
| call batch.batch as batch { | |||
| input: | |||
| sample_id = sample_id, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| tumor_bam = tumor_bam, | |||
| tumor_bai = tumor_bai, | |||
| normal_bam = normal_bam, | |||
| normal_bai = normal_bai, | |||
| bed = bed, | |||
| access_bed = access.access_bed, | |||
| docker = docker, | |||
| cluster = cluster, | |||
| disk_size = disk_size | |||
| } | |||
| call export.export as export { | |||
| input: | |||
| result = batch.result, | |||
| docker = docker, | |||
| cluster = cluster, | |||
| disk_size = disk_size | |||
| } | |||
| } | |||
Завантаження…