
 junshang
6e856f691e
hg38 to hg_ref
junshang
6e856f691e
hg38 to hg_ref
|
il y a 4 ans | |
|---|---|---|
| assest | il y a 4 ans | |
| tasks | il y a 4 ans | |
| README.md | il y a 4 ans | |
| defaults | il y a 4 ans | |
| inputs | il y a 4 ans | |
| workflow.wdl | il y a 4 ans | |
README.md
CNVkit
Author: Yaqing Liu
E-mail: yaqing.liu@outlook.com
CNVkit is a Python library and command-line software toolkit to infer and visualize copy number from high-throughput DNA sequencing data. It is designed for use with hybrid capture, including both whole-exome and custom target panels, and short-read sequencing platforms such as Illumina and Ion Torrent.
Official document: https://cnvkit.readthedocs.io/en/stable/index.html
Install
# activate choppy environment
open-choppy-env
# install app
choppy install YaqingLiu/CNVkit
Input
Please note that the input file of this APP is in the JSON format instead of CSV.
{
"tumor_bam": [
"oss://choppy-cromwell-result/...bam",
"oss://choppy-cromwell-result/...bam",
"oss://choppy-cromwell-result/...bam"
],
"tumor_bai": [
"oss://choppy-cromwell-result/...bai",
"oss://choppy-cromwell-result/...bai",
"oss://choppy-cromwell-result/...bai"
],
"normal_bam": [
"oss://choppy-cromwell-result/...bam",
"oss://choppy-cromwell-result/...bam",
"oss://choppy-cromwell-result/...bam"
],
"normal_bai": [
"oss://choppy-cromwell-result/...bai",
"oss://choppy-cromwell-result/...bai",
"oss://choppy-cromwell-result/...bai"
],
"sample_id": "...",
"method": "...",
"reference": "..." # this parameter is optional
}
Submit your task
# choppy batch
chopppy batch YaqingLiu/CNVkit-latest samples.json -p project_name
Copy number calling pipeline
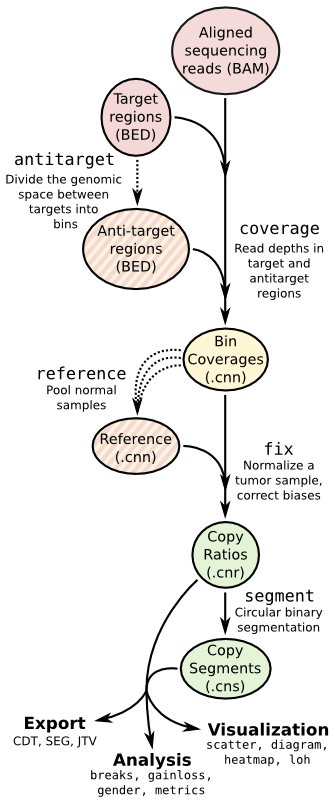
Note
-m {hybrid,amplicon,wgs}, --seq-method {hybrid,amplicon,wgs}, --method {hybrid,amplicon,wgs}
Sequencing assay type: hybridization capture (‘hybrid’), targeted amplicon sequencing (‘amplicon’), or whole genome sequencing (‘wgs’). Determines whether and how to use antitarget bins.
sequencing-accessible regions
Many fully sequenced genomes, including the human genome, contain large regions of DNA that are inaccessable to sequencing. (These are mainly the centromeres, telomeres, and highly repetitive regions.) In the FASTA genome sequence these regions are filled in with large stretches of N characters. These regions cannot be mapped by resequencing, so we can avoid them when calculating the antitarget locations by passing the locations of the accessible sequence regions with the -g or --access option.
To use CNVkit on amplicon sequencing data instead of hybrid capture – although this is not recommended – you can exclude all off-target regions from the analysis by passing the target BED file as the “access” file as well:
cnvkit.py batch ... -t Tiled.bed -g Tiled.bed ...
This results in empty ”.antitarget.cnn” files which CNVkit will handle safely from version 0.3.4 onward. However, this approach does not collect any copy number information between targeted regions, so it should only be used if you have in fact prepared your samples with a targeted amplicon sequencing protocol.
To reuse an existing reference or create a new:
-r REFERENCE, --reference REFERENCE
Copy number reference file (.cnn).
--output-reference FILENAME
Output filename/path for the new reference file being created. (If given, ignores the -o/--output-dir option and will write the file to the given path. Otherwise, “reference.cnn” will be created in the current directory or specified output directory.)
--annotate
The gene annotations file (refFlat.txt) is useful to apply gene names to your baits BED file, if the BED file does not already have short, informative names for each bait interval. This file can be used in the next step. If the BED looks like this:
chr1 1508981 1509154 SSU72
chr1 2407978 2408183 PLCH2
chr1 2409866 2410095 PLCH2
Then you don’t need refFlat.txt.
index files
If you’ve prebuilt the index file (.bai, .fai), make sure its timestamp is later than the BAM file’s and fa’s.
CNVkit will automatically index the file if needed – that is, if the .bai/.fa file is missing, or if the timestamp of the .bai/.fa file is older than that of the corresponding .bam/.fa file.
_-s min_gapsize
Minimum gap size between accessible sequence regions. Regions separated by less than this distance will be joined together. [Default: 5000]
segment method Segmentation methods The following segmentation algorithms can be specified with the -m option:
cbs – the default, circular binary segmentation (CBS). This method performed best in our benchmarking on mid-size target panels and exomes. Requires the R package DNAcopy. flasso – Fused Lasso, reported by some users to perform best on exomes, whole genomes, and some target panels. Sometimes faster than CBS, but the current implementation cannot be parallelized over multiple CPUs. Beyond identifying breakpoints, additionally performs significance testing to distinguish CNAs from regions of neutral copy number, so large swathes of the output may have log2 values of exactly 0. Requires the R package cghFLasso.
haar – a pure-Python implementation of HaarSeg, a wavelet-based method. Very fast and performs reasonably well on small panels, but tends to over-segment large datasets. hmm (experimental) – a 3-state Hidden Markov Model suitable for most samples. Faster than CBS, and slower but more accurate than Haar. Requires the Python package hmmlearn, as do the next two methods.
hmm-tumor (experimental) – a 5-state HMM suitable for finer-grained segmentation of good-quality tumor samples. In particular, this method can detect focal amplifications within a larger-scale, smaller-amplitude copy number gain, or focal deep deletions within a larger-scale hemizygous loss. Training this model takes a bit more CPU time than the simpler hmm method.
hmm-germline (experimental) – a 3-state HMM with fixed amplitude for the loss, neutral, and gain states corresponding to absolute copy numbers of 1, 2, and 3. Suitable for germline samples and single-cell sequencing of samples with mostly-diploid genomes that are not overly aneuploid.
none – simply calculate the weighted mean log2 value of each chromosome arm. Useful for testing or debugging, or as a baseline for benchmarking other methods. The first two methods use R internally, and to use them you will need to have R and the R package dependencies installed (i.e. DNAcopy, cghFLasso). If you installed CNVkit with conda as recommended, these should have been installed for you automatically. If you installed the R packages in a nonstandard or non-default location, you can specify the location of the right Rscript executable you want to use with --rscript-path.
The HMM methods hmm, hmm-tumor and hmm-germline were introduced provisionally in CNVkit v.0.9.2, and may change in future releases.
Output
- *.cnn/cns of each sample.
- A whole-genome copy ratio profile as a PDF scatter plot.
- An ideogram of copy ratios on chromosomes as a PDF.
- A segment file which can be imported into IGV.