
 LUYAO REN
753b1c1fcc
hardfiltration
LUYAO REN
753b1c1fcc
hardfiltration
|
6 лет назад | |
|---|---|---|
| pictures | 6 лет назад | |
| tasks | 6 лет назад | |
| README.md | 6 лет назад | |
| inputs | 6 лет назад | |
| workflow.wdl | 6 лет назад | |
README.md
RNAseq variant calling
RNAseq variant calling pipeline:
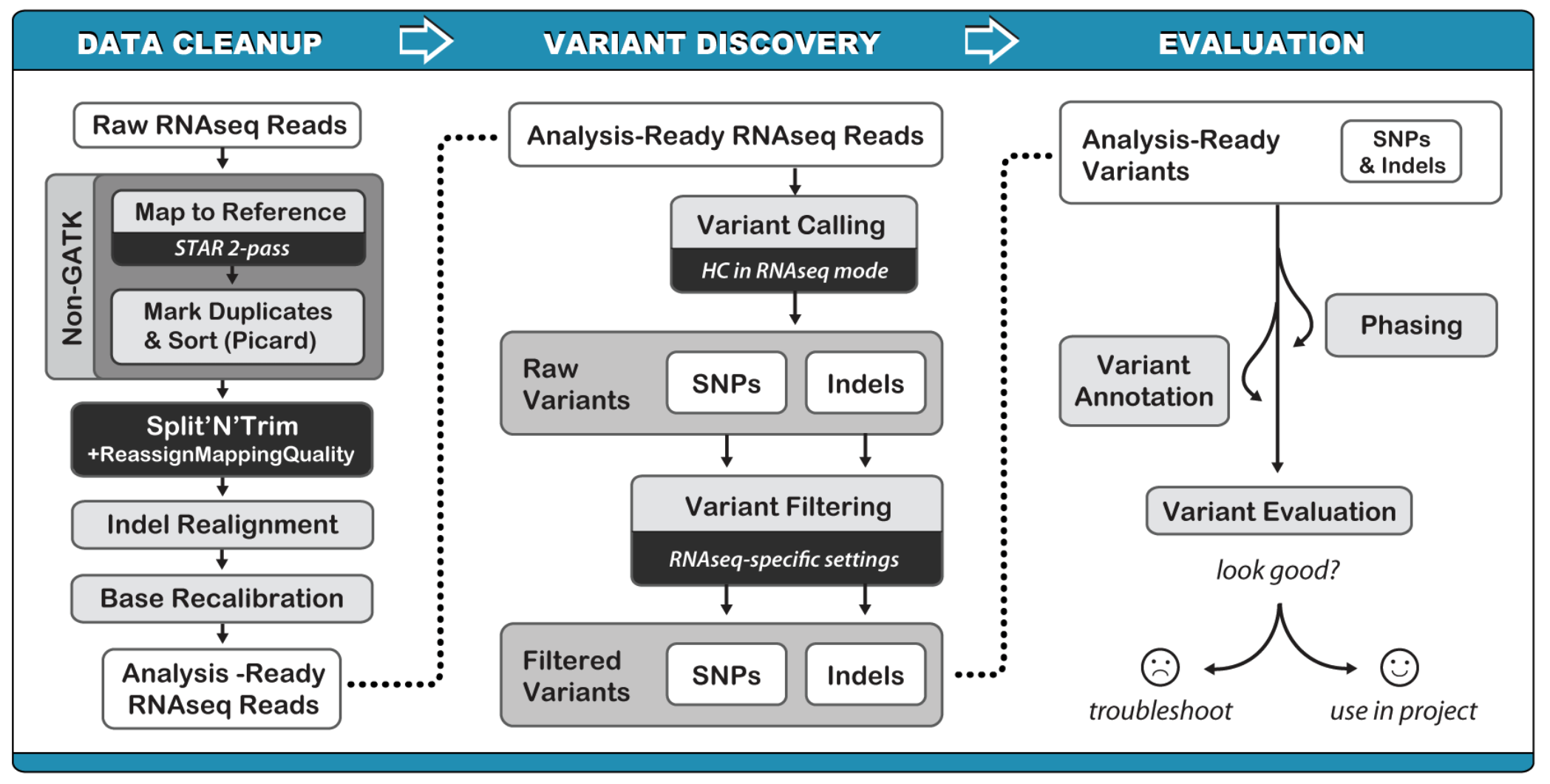
(1) Mapping to the reference genome (STAR)
Aligning RNAseq data to a reference genome is complicates by RNA splicing, GATK RNAseq variant calling best practice recommends STAR.
Generate genome index files:
The STAR you use
genomeDir=/path/to/GRCh38
mkdir $genomeDir
STAR --runMode genomeGenerate\
--genomeDir $genomeDir\
--genomeFastaFiles hg19.fa\
--runThreadN <n>
Mapping reads to the genome:
The author recommend running STAR in the 2-pass mode for the most sensitive novel junction discovery. It does not increase the number of detected novel junctions, but allows to detect more splices reads mapping to novel junctions. The basic idea is to run 1st pass of STAR mapping with the usual paraments, then collect the junctions detected inthe first pass, and use them as “annotated” junctions for the 2nd pass mapping
# 1. 1st pass
runDir=/path/to/1pass
mkdir $runDir
cd $runDir
STAR --genomeDir $genomeDir --readFilesIn mate1.fq mate2.fq --runThreadN <n>
# For the 2-pass STAR, a new index is then created using splice junstion information contained inthe file SJ.out.tab from the first pass
genomeDir=/path/to/hg19_2pass
mkdir $genomeDir
STAR --runMode genomeGenerate --genomeDir $genomeDir --genomeFastaFiles hg19.fa \
--sjdbFileChrStartEnd /path/to/1pass/SJ.out.tab --sjdbOverhang 75 --runThreadN <n>
# 3. 2nd pass
runDir=/path/to/2pass
mkdir $runDir
cd $runDir
STAR --genomeDir $genomeDir --readFilesIn mate1.fq mate2.fq --runThreadN <n>
(2) Convert alignment output SAM to BAM, and add read groups (picard) and index bam (samtools)
java -jar picard.jar AddOrReplaceReadGroups I=star_output.sam O=rg_added_sorted.bam SO=coordinate RGID=id RGLB=library RGPL=platform RGPU=machine RGSM=sample
# index bam and get .bai
samtools index rg_added_sorted.bam
(3) Remove deplicates
sentieon driver -t NUMBER_THREADS -i SORTED_BAM \
--algo LocusCollector --fun score_info SCORE.gz
sentieon driver -t NUMBER_THREADS -i SORTED_BAM \
--algo Dedup --rmdup --score_info SCORE.gz \
--metrics DEDUP_METRIC_TXT DEDUPED_BAM
(4) Split reads at junction
This step slits the RNA reads into exon segments by getting rid of Ns while maintaining grouping information, and hard-clips any sequences overhanging into the intron regions. Additionally, the step will reassign the mapping qualities from STAR to be consistent with what is expected in subsequent steps by converting from quality 255 to 60.
sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \
--algo RNASplitReadsAtJunction --reassign_mapq 255:60 SPLIT_BAM
(5) Base quality score recalibration
sentieon driver -r $fasta -t $nt -i SPLIT_BAM --algo QualCal -k $dbsnp -k $known_Mills_indels -k $known_1000G_indels recal_data.table
sentieon driver -r $fasta -t $nt -i SPLIT_BAM -q recal_data.table --algo QualCal -k $dbsnp -k $known_Mills_indels -k $known_1000G_indels recal_data.table.post
sentieon driver -t $nt --algo QualCal --plot --before recal_data.table --after recal_data.table.post recal.csv
sentieon plot QualCal -o recal_plots.pdf recal.csv
(6) Variant calling
sentieon driver -t NUMBER_THREADS -r REFERENCE -i SPLIT_BAM \
-q RECAL_DATA.TABLE --algo Haplotyper --trim_soft_clip \
--call_conf 20 --emit_conf 20 [-d dbSNP] VARIANT_VCF
(7) Variant Filtering
Hard filtration is applied, for GATK do not yet have the RNAseq training/truth resources that would be needed to run VQSR.
-window 35 -cluster 2Remove clusters of at least 3 SNPs that are within a window of 35 bases between themFS > 30.0Fisher Strand valuesQD < 2.0Qual By Depth values
# GATK3, if you are using gatk4, please pay attention to the new arguments
java -jar GenomeAnalysisTK.jar -T VariantFiltration -R hg_19.fasta -V input.vcf -window 35 -cluster 3 -filterName FS -filter "FS > 30.0" -filterName QD -filter "QD < 2.0" -o output.vcf
Per sample running time
Less than 4 hours
Settings:
- Disksize 500
- Cluster OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc
| Module | Time |
|---|---|
| Mapping | 1h20min |
| SamToBam | 1h |
| indexBam | 5min |
| Dedup | 13min |
| SplitReads | 6min |
| BQSR | 21min |
| Haplotyper | 15min |
| Hardfiltration | 3min |
Reference
- GATK best practice https://software.broadinstitute.org/gatk/documentation/article.php?id=3891
- Sentieon manual https://support.sentieon.com/manual/RNA_call/rna/
- STAR github https://github.com/alexdobin/STAR
- Picard github https://github.com/broadinstitute/picard
- Picard manual https://broadinstitute.github.io/picard/
- samtools manual http://www.htslib.org/doc/samtools.html