|
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465666768697071727374 |
- # CNVkit
-
- > Author: Yaqing Liu
- >
- > E-mail: yaqing.liu@outlook.com
- >
-
- CNVkit is a Python library and command-line software toolkit to infer and visualize copy number from high-throughput DNA sequencing data. It is designed for use with hybrid capture, including both whole-exome and custom target panels, and short-read sequencing platforms such as Illumina and Ion Torrent.
-
- Official document: https://cnvkit.readthedocs.io/en/stable/index.html
- ## Install
-
- ```
- # activate choppy environment
- open-choppy-env
- # install app
- choppy install YaqingLiu/CNVkit
- ```
-
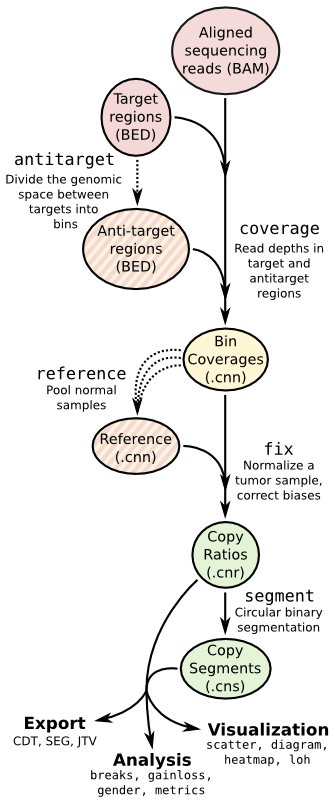
- ## Copy number calling pipeline
- 
-
- ## Input
- ```json
- {
- "tumor_bam": [
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam"
- ],
- "tumor_bai": [
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai"
- ],
- "normal_bam": [
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam"
- ],
- "normal_bai": [
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai"
- ],
- "sample_id": "...",
- "method": "...",
- "reference": "..." # this parameter is optional
- }
- ```
- ***Note***
- ```shell
- -m {hybrid,amplicon,wgs}, --seq-method {hybrid,amplicon,wgs}, --method {hybrid,amplicon,wgs}
- Sequencing assay type: hybridization capture
- ('hybrid'), targeted amplicon sequencing
- ('amplicon'), or whole genome sequencing ('wgs').
- Determines whether and how to use antitarget bins.
-
- To reuse an existing reference or create a new :
- -r REFERENCE, --reference REFERENCE
- Copy number reference file (.cnn).
- --output-reference FILENAME
- Output filename/path for the new reference file
- being created. (If given, ignores the
- -o/--output-dir option and will write the file to
- the given path. Otherwise, "reference.cnn" will
- be created in the current directory or specified
- output directory.)
- ```
- ## Output
- 1. *.cnn/cns of each sample.
- 2. A whole-genome copy ratio profile as a PDF scatter plot.
- 3. An ideogram of copy ratios on chromosomes as a PDF.
- 4. A segment file which can be imported into IGV.
|

