|
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475 |
- # CNVkit
-
- > Author: Yaqing Liu
- >
- > E-mail: yaqing.liu@outlook.com
- >
-
- CNVkit is a Python library and command-line software toolkit to infer and visualize copy number from high-throughput DNA sequencing data. It is designed for use with hybrid capture, including both whole-exome and custom target panels, and short-read sequencing platforms such as Illumina and Ion Torrent.
-
- Official document: https://cnvkit.readthedocs.io/en/stable/index.html
- ## Install
-
- ```
- # activate choppy environment
- open-choppy-env
- # install app
- choppy install YaqingLiu/CNVkit
- ```
-
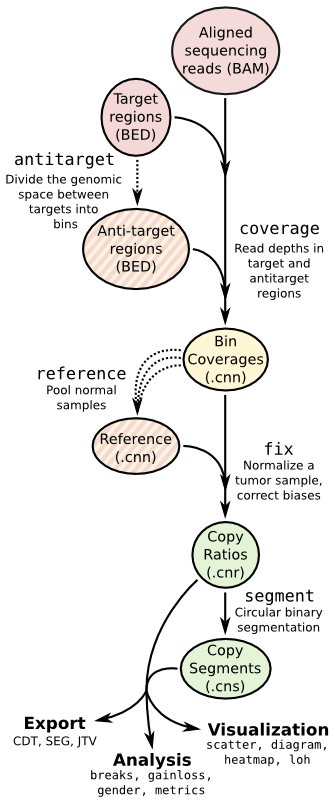
- ## Copy number calling pipeline
- 
-
- ## Input
- ```json
- {
- "tumor_bam": [
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam"
- ],
- "tumor_bai": [
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai"
- ],
- "normal_bam": [
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam",
- "oss://choppy-cromwell-result/...bam"
- ],
- "normal_bai": [
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai",
- "oss://choppy-cromwell-result/...bai"
- ],
- "sample_id": "...",
- "method": "...",
- "reference": "..." # this parameter is optional
- }
- ```
- ***Note***
- ```shell
- -m {hybrid,amplicon,wgs}, --seq-method {hybrid,amplicon,wgs}, --method {hybrid,amplicon,wgs}
- Sequencing assay type: hybridization capture ('hybrid'), targeted amplicon sequencing ('amplicon'), or whole genome sequencing ('wgs').
- Determines whether and how to use antitarget bins.
-
- To reuse an existing reference or create a new :
- -r REFERENCE, --reference REFERENCE
- Copy number reference file (.cnn).
- --output-reference FILENAME
- Output filename/path for the new reference file being created. (If given, ignores the -o/--output-dir option and will write the file to the given path. Otherwise, "reference.cnn" will be created in the current directory or specified output directory.)
-
- --annotate
- The gene annotations file (refFlat.txt) is useful to apply gene names to your baits BED file, if the BED file does not already have short, informative names for each bait interval. This file can be used in the next step.
- If the BED looks like this:
- > chr1 1508981 1509154 SSU72
- > chr1 2407978 2408183 PLCH2
- > chr1 2409866 2410095 PLCH2
- Then you don’t need refFlat.txt.
- ```
- ## Output
- 1. *.cnn/cns of each sample.
- 2. A whole-genome copy ratio profile as a PDF scatter plot.
- 3. An ideogram of copy ratios on chromosomes as a PDF.
- 4. A segment file which can be imported into IGV.
|

