
8.2KB
中华家系1号标准物质室间质评报告系统分析流程
Author: Run Luyao
E-mail:18110700050@fudan.edu.cn
Git: http://choppy.3steps.cn/renluyao/Quality_control.git
Last Updates: 30/8/2019
安装指南
# 激活choppy环境
source activate choppy
# 安装app
choppy install renluyao/Quality_control
App概述——中华家系1号标准物质介绍
建立高通量全基因组测序的生物计量和质量控制关键技术体系,是保障测序数据跨技术平台、跨实验室可比较、相关研究结果可重复、数据可共享的重要关键共性技术。建立国家基因组标准物质和基准数据集,突破基因组学的生物计量技术,是将测序技术转化成临床应用的重要环节与必经之路,目前国际上尚属空白。中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制了人源中华家系1号基因组标准物质(Quartet,一套4个样本,编号分别为LCL5,LCL6,LCL7,LCL8,其中LCL5和LCL6为同卵双胞胎女儿,LCL7为父亲,LCL8为母亲),以及相应的全基因组测序序列基准数据集(“量值”),为衡量基因序列检测准确与否提供一把“标尺”,成为保障基因测序数据可靠性的国家基准。人源中华家系1号基因组标准物质来源于泰州队列同卵双生双胞胎家庭,从遗传结构上体现了我国南北交界的人群结构特征,同时家系的设计也为“量值”的确定提供了遗传学依据。
中华家系1号DNA标准物质的标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和77.9-78.1%的高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。
该Quality_control APP用于全基因组测序(whole-genome sequencing,WGS)数据的质量评估,包括原始数据质控、比对数据质控和突变检出数据质控。
流程与参数
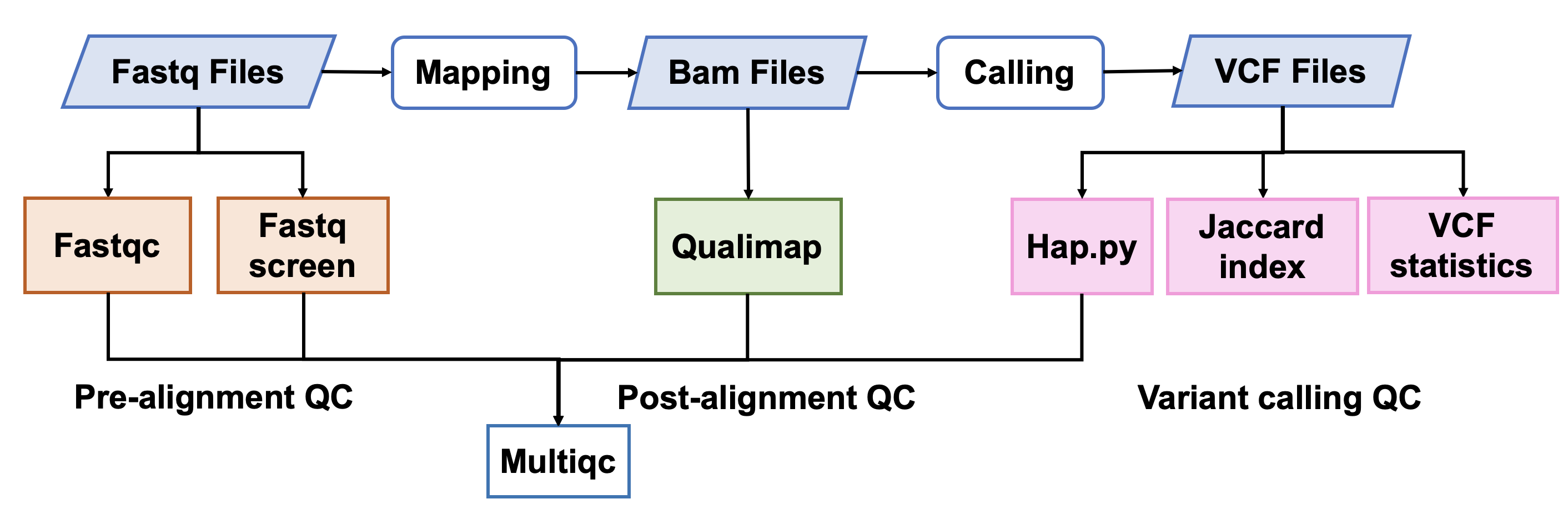
1. 原始数据质量控制
Fastqc
FastQC是一个常用的测序原始数据的质控软件,主要包括12个模块,具体请参考Fastqc模块详情。
fastqc -t <threads> -o <output_directory> <fastq_file>
Fastq Screen
Fastq Screen是检测测序原始数据中是否引⼊入其他物种,或是接头引物等污染,⽐比如,如果测序样本 是⼈人类,我们期望99%以上的reads匹配到⼈人类基因组,10%左右的reads匹配到与⼈人类基因组同源性 较⾼高的⼩小⿏鼠上。如果有过多的reads匹配到Ecoli或者Yeast,要考虑是否在培养细胞的时候细胞系被污 染,或者建库时⽂文库被污染。
fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file>
--conf conifg 文件主要输入了多个物种的fasta文件地址,可根据自己自己的需求下载其他物种的fasta文件加入分析
--top一般不需要对整个fastq文件进行检索,取前100000行
``
2. 比对后数据质量控制
Qualimap
qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G
3. 突变检出数据质量控制
Hap.py
hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename>
Jaccard index
rtg vcfeval -b <one_vcf> -c <another_vcf> -o <output_directory> -t <sdf_file>
VCF statistics
rtg vcfstats <vcf_file>
App输入变量与输入文件
准备inputSamplesFIle (tsv格式)
#fastq_read1 #fastq_read2 #bam #bai #vcf #sample_mark
App输出文件
结果展示与解读
GSEA结果解读示例:
1. Enrichment score(ES)
ES是GSEA最初的结果,反应全部杂交data排序后,在此序列top或bottom富集的程度。 ES原理:扫描排序序列,当出现一个功能集中的gene时,增加ES值,反之减少ES值,所以ES是个动态值。最终ES的确定是讲杂交数据排序序列所在位置定义为0,ES值定义为距离排序序列的最大偏差.
- ES为正,表示某一功能gene集富集在排序序列前方
- ES为负,表示某一功能gene集富集在排序序列后方。 图中的最高点为此通路的ES值,中间表示杂交数据的排序序列。竖线表示此通路中出现的芯片数据集中的gene。
2. NES
由于ES是根据分析的数据集中的gene是否在一个功能gene set中出现来计算的,但各个功能gene set中包含的gene数目不同,且不同功能gene set与data之间的相关性也不同,因此,比较data set在不同功能gene set中的富集程度要对ES进行标准化处理,也就是NES NES=某一功能gene set的ES/数据集所有随机组合得到的ES平均值 NES是主要的统计量。
3. FDR
NES确定后,判断其中可能包含的错误阳性发现率。FDR=25%意味着对此NES的确定,4次可能错 1次。GSEA结果中,高亮显示FDR<25%的富集set。因为从这些功能gene中最可能产生有意义的假设,促进进一步研究。大多数情况下,选FDR<25%是合适的,但是,假如分析的芯片data set较少,选择的是探针随机组合而不是表型组合,若p不严格,那么应该选FDR<5%。一般而言,NES绝对值越大,FDR值就越小,说明富集程度高,结果可靠。
4. 名义p值 nominal p-value
描述的是针对某一功能gene子集得到的富集得分的统计显著性,显然,p越小,富集性越好。
以上4个参数中,只有FDR进行了功能gene子集大小和多重假设检验矫正,而p值没有,因此,如果结果中有一个高度富集的功能gene子集,而其有很小的名义p-value和大的FDR意味着富集并不显著。
我的一个具体结果解读:
92/681 gene sets are upregulated in PH 0 gene sets are significantly enriched at FDR<25% 1 gene sets are significantly enriched at n p-value <1% 1 gene sets are significantly enriched at n p-value <5%
在选择的BP中,有681个gene sets,92个PH中上调,其中75%的正确率支持0条子集上调,1个BP的gene表达上调名义p值<0.01。总体结果并不理想。
5. 备注
GSEA富集结果太少说明:
无gene set被富集。可能是因为分析的样本太少,关注的生物信息太微弱,或正在分析的功能集不能很好代表你所关心的生物过程,但仍然可以看下top ranked gene sets,这些信息可能会为你的假说提供微弱的证据。当然也可以尝试考虑分析其他gene sets,或增加samples
GSEA富集结果太多说明:
太多的功能子集被富集了。可能是因为很多的gene sets代表同一生物信号,这可以在gene sets中查看leading edge sbusets来查看。或者也可以查看具体区别进行加工,比如samples来自不同labs,操作者不一样等。
CHANGELOG
Version 1.0 - Auguest 30, 2019
- 完成PGx常规质控流程的choppy APP
FAQ
1. RNAseq和甲基化的质控流程?
可查询multiqc支持的质控模块 https://multiqc.info/docs/#multiqc-modules
RNAseq和甲基化的质控流程待完善
2. 如果样本没有技术重复,该APP中的inputJIpiarsFile是怎么输入的?
在Version 1.0中暂时还没有考虑没有技术重复的问题,可输入姐妹、父母、父女、母女的配对,计算同卵双胞胎、亲属关系和陌生人之间基因突变位点的一致性。