
 LUYAO REN
6 年前
LUYAO REN
6 年前
当前提交
e976c81561
共有 17 个文件被更改,包括 520 次插入 和 0 次删除
+ 33
- 0
README.md
查看文件
| # 中华家系1号标准物质室间质评报告系统分析流程 | |||||
| ###1. 中华家系1号标准物质介绍 | |||||
| ###2. 分析流程图 | |||||
| ###3. 各模块运行流程 | |||||
| 准备inputSamplesFIle (tsv格式) | |||||
| ```bash | |||||
| #fastq_read1 #fastq_read2 #bam #bai #vcf #sample_mark | |||||
| ``` | |||||
| (1) fastqc | |||||
| (2) fastqscreen | |||||
| (3) qualimap | |||||
| (4) benchmarking ([hap.py](<https://github.com/Illumina/hap.py/blob/master/doc/happy.md>)) | |||||
| (5) multiqc | |||||
+ 152
- 0
Report.md
查看文件
| # 基于中华家系1号的全基因组数据质量控制报告 | |||||
| ###1. 基本信息 | |||||
| **机构名称:**复旦大学 | |||||
| **建库方法:**TruSeq | |||||
| **测序仪器:**Illumina Novaseq | |||||
| **Read长度:**双端150bp | |||||
| **参考基因组:**GRCh38 | |||||
| **回收数据:** | |||||
| | 样本 | Fastq | Bam | VCF | | |||||
| | :------: | :---: | :--: | :--: | | |||||
| | Fudan D5 | 3 | 3 | 3 | | |||||
| | Fudan D6 | 3 | 3 | 3 | | |||||
| | Fudan F7 | 3 | 3 | 3 | | |||||
| | Fudan M8 | 3 | 3 | 3 | | |||||
| **分析流程:** | |||||
| | 步骤 | 软件 | 版本号 | | |||||
| | :---------------: | :---------: | :----: | | |||||
| | Preprocessing | Trimmomatic | | | |||||
| | Mapping | BWA-MEM | | | |||||
| | Remove Duplicates | Picard | | | |||||
| | BQSR | GATK | | | |||||
| | Calling | Haplotyper | | | |||||
| | Filtration | VQSR | | | |||||
| ###2. 原始数据质量评估 | |||||
| | 项目名称 | Fudan D5 | Fudan D6 | Fudan F7 | Fudan M8 | ... | 参考值 | | |||||
| | :-------------------: | :------: | :------: | :------: | :------: | :--: | ------ | | |||||
| | 原始数据量(Million) | | | | | | | | |||||
| | Reads重复率(%) | | | | | | | | |||||
| | 原始数据测序深度 | | | | | | | | |||||
| | 碱基质量 | | | | | | | | |||||
| | ATGC含量 (%) | | | | | | | | |||||
| | GC含量分布(%) | | | | | | | | |||||
| | 重复序列(%) | | | | | | | | |||||
| | 接头序列含量(%) | | | | | | | | |||||
| | 污染物 | | | | | | | | |||||
| ###3. 比对后数据质量评估 | |||||
| | 项目名称 | Fudan D5 | Fudan D6 | Fudan F7 | Fudan M8 | ... | 参考值 | | |||||
| | :--------------------: | :------: | :------: | -------- | -------- | :--: | ------ | | |||||
| | 比对率(%) | | | | | | | | |||||
| | 高质量Reads比对率(%) | | | | | | | | |||||
| | 错配率(%) | | | | | | | | |||||
| | 去重后测序深度 | | | | | | | | |||||
| | Insert size (bp) | | | | | | | | |||||
| ###4. 突变数据质量评估 | |||||
| ####(1)突变统计 | |||||
| | Sample | Fudan D5 | Fudan D6 | Fudan F7 | Fudan M8 | ... | | |||||
| | :---------------------------: | :------: | :------: | :------: | :------: | :--: | | |||||
| | 突变总数 | | | | | | | |||||
| | SNVs | | | | | | | |||||
| | Insertions | | | | | | | |||||
| | Deletions | | | | | | | |||||
| | SNV Transitions/Transversions | | | | | | | |||||
| | Total Het/Hom ratio | | | | | | | |||||
| | SNV Het/Hom ratio | | | | | | | |||||
| | Insertions Het/Hom ratio | | | | | | | |||||
| | Deletions Het/Hom ratio | | | | | | | |||||
| ####(2)技术重复一致性 | |||||
| | 技术重复的一致性 | Fudan D5 | Fudan D6 | Fudan F7 | Fudan M8 | ... | | |||||
| | ---------------- | -------- | -------- | -------- | -------- | ---- | | |||||
| | SNVs | | | | | | | |||||
| | Indels | | | | | | | |||||
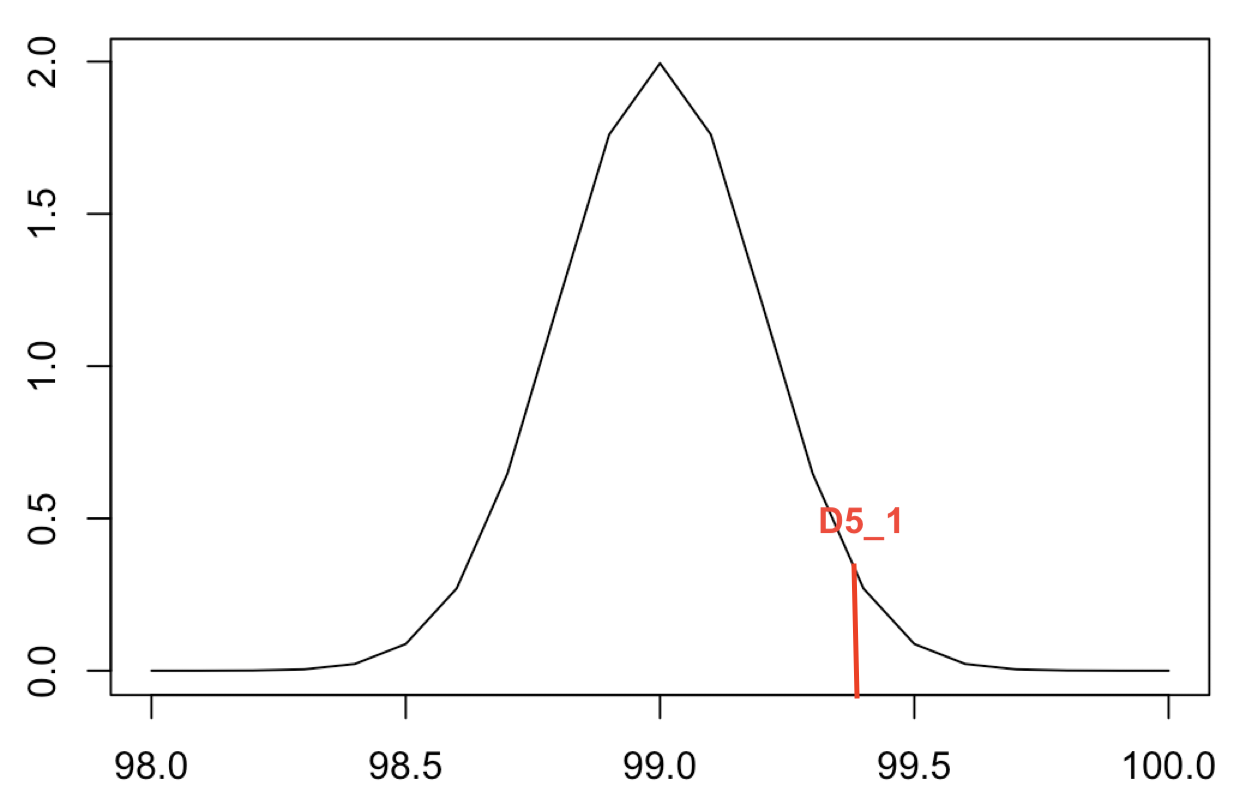
|  | |||||
| ####(3)突变检测准确性(Presicion, recall, F1-score) | |||||
| | 基因环境 | 类型 | Fudan D5 | Fudan D6 | Fudan F7 | Fudan M8 | | |||||
| | ------------------------------------------------------------ | ----- | -------- | -------- | -------- | -------- | | |||||
| | All | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Not in homopolymers or TRs | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | In homopolymers or TRs | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | GC content | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Tier 1 (supported by all replicates, sequencing sites, platforms and bioinformatic pipelines) | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Tier 2 (supported by majority of replicates, sequencing sites, platforms and bioinformatic pipelines ) | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Tier 3 (supported by only one platform and multiple bioinformatic pipelines) | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | In high confidence bed | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Not in high confidence bed | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | In structural variantion region | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | Clinical relevant mutations | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
| | De novo | SNV | | | | | | |||||
| | | Indel | | | | | | |||||
|  | |||||
| #### 下载 | |||||
| **SNV** | |||||
| - True Positive | |||||
| - True Negative | |||||
| - False Positive | |||||
| - False Negative | |||||
| **INDEL** | |||||
| - True Positive | |||||
| - True Negative | |||||
| - False Positive | |||||
| - False Negative | |||||
| ### 5. 附录 | |||||
| **(1) 中华家系1号DNA标准物质介绍** | |||||
| 中华家系1号全基因组DNA标准物质由中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制。候选物来自同卵双胞胎家庭的永生化B淋巴母细胞系,志愿者选自复旦大学泰州队列,泰州地处我国南北交界,代表了中国人群典型的遗传结构特征。由于同卵双生双胞胎家庭的家系设计,可以通过孟德尔遗传定律进一步排除标称值确定过程中的可能错误。同时,中华家系1号转录组、蛋白质组和代谢物组的标准物质也在逐步研制中,通过多组学数据的整合分析可为标称值的确定提供了另一层面的生物学依据。 | |||||
| 中华家系1号DNA标准物质的标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和77.9-78.1%的高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。 | |||||
|  | |||||
| **(2) 数据分析方法与流程** | |||||
| 数据分析流程如下图所示,利用Sentieon进行数据比对、比对后校正、突变分析和过滤,利用FastQC、Qualimap、MultiQC、RTGtools、R和in-house script进行数据的质量控制和评估。采用choppy分析调度和完成报告。 | |||||
|  | |||||
| ###6. 声明 | |||||
| 本质量检测报告,仅适用于此次实验测试数据,不代表对测序公司业务水平的评估。本质量检测报告,仅用于科学项目研究,请勿用于临床或商业。任何单位或个人因使用此检测报告结果造成的任何利益或损失(包括直接和间接损失),本单位不承担任何经济和法律责任。 | |||||
+ 94
- 0
fastq_screen.conf
查看文件
| # This is an example configuration file for FastQ Screen | |||||
| ############################ | |||||
| ## Bowtie, Bowtie 2 or BWA # | |||||
| ############################ | |||||
| ## If the Bowtie, Bowtie 2 or BWA binary is not in your PATH, you can set | |||||
| ## this value to tell the program where to find your chosen aligner. Uncomment | |||||
| ## the relevant line below and set the appropriate location. Please note, | |||||
| ## this path should INCLUDE the executable filename. | |||||
| #BOWTIE /usr/local/bin/bowtie/bowtie | |||||
| #BOWTIE2 /usr/local/bowtie2/bowtie2 | |||||
| #BWA /usr/local/bwa/bwa | |||||
| ############################################ | |||||
| ## Bismark (for bisulfite sequencing only) # | |||||
| ############################################ | |||||
| ## If the Bismark binary is not in your PATH then you can set this value to | |||||
| ## tell the program where to find it. Uncomment the line below and set the | |||||
| ## appropriate location. Please note, this path should INCLUDE the executable | |||||
| ## filename. | |||||
| #BISMARK /usr/local/bin/bismark/bismark | |||||
| ############ | |||||
| ## Threads # | |||||
| ############ | |||||
| ## Genome aligners can be made to run across multiple CPU cores to speed up | |||||
| ## searches. Set this value to the number of cores you want for mapping reads. | |||||
| THREADS 32 | |||||
| ############## | |||||
| ## DATABASES # | |||||
| ############## | |||||
| ## This section enables you to configure multiple genomes databases (aligner index | |||||
| ## files) to search against in your screen. For each genome you need to provide a | |||||
| ## database name (which can't contain spaces) and the location of the aligner index | |||||
| ## files. | |||||
| ## | |||||
| ## The path to the index files SHOULD INCLUDE THE BASENAME of the index, e.g: | |||||
| ## /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 | |||||
| ## Thus, the index files (Homo_sapiens.GRCh37.1.bt2, Homo_sapiens.GRCh37.2.bt2, etc.) | |||||
| ## are found in a folder named 'GRCh37'. | |||||
| ## | |||||
| ## If, for example, the Bowtie, Bowtie2 and BWA indices of a given genome reside in | |||||
| ## the SAME FOLDER, a SINLGE path may be provided to ALL the of indices. The index | |||||
| ## used will be the one compatible with the chosen aligner (as specified using the | |||||
| ## --aligner flag). | |||||
| ## | |||||
| ## The entries shown below are only suggested examples, you can add as many DATABASE | |||||
| ## sections as required, and you can comment out or remove as many of the existing | |||||
| ## entries as desired. We suggest including genomes and sequences that may be sources | |||||
| ## of contamination either because they where run on your sequencer previously, or may | |||||
| ## have contaminated your sample during the library preparation step. | |||||
| ## | |||||
| ## Human - sequences available from | |||||
| ## ftp://ftp.ensembl.org/pub/current/fasta/homo_sapiens/dna/ | |||||
| #DATABASE Human /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 | |||||
| ## | |||||
| ## Mouse - sequence available from | |||||
| ## ftp://ftp.ensembl.org/pub/current/fasta/mus_musculus/dna/ | |||||
| #DATABASE Mouse /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37 | |||||
| ## | |||||
| ## Ecoli- sequence available from EMBL accession U00096.2 | |||||
| #DATABASE Ecoli /data/public/Genomes/Ecoli/Ecoli | |||||
| ## | |||||
| ## PhiX - sequence available from Refseq accession NC_001422.1 | |||||
| #DATABASE PhiX /data/public/Genomes/PhiX/phi_plus_SNPs | |||||
| ## | |||||
| ## Adapters - sequence derived from the FastQC contaminats file found at: www.bioinformatics.babraham.ac.uk/projects/fastqc | |||||
| #DATABASE Adapters /data/public/Genomes/Contaminants/Contaminants | |||||
| ## | |||||
| ## Vector - Sequence taken from the UniVec database | |||||
| ## http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html | |||||
| #DATABASE Vectors /data/public/Genomes/Vectors/Vectors | |||||
| DATABASE Human /cromwell_inputs/*/fastq_screen_reference/genome | |||||
| DATABASE Mouse /cromwell_inputs/*/fastq_screen_reference/mouse | |||||
| DATABASE ERCC /cromwell_inputs/*/fastq_screen_reference/ERCC | |||||
| DATABASE EColi /cromwell_inputs/*/fastq_screen_reference/ecoli | |||||
| DATABASE Adapter /cromwell_inputs/*/fastq_screen_reference/adapters | |||||
| DATABASE Vector /cromwell_inputs/*/fastq_screen_reference/vector | |||||
| DATABASE rRNA /cromwell_inputs/*/fastq_screen_reference/rRNARef | |||||
| DATABASE Virus /cromwell_inputs/*/fastq_screen_reference/viral | |||||
| DATABASE Yeast /cromwell_inputs/*/fastq_screen_reference/GCF_000146045.2_R64_genomic_modify | |||||
| DATABASE Mitoch /cromwell_inputs/*/fastq_screen_reference/human_mitoch/Human_mitoch | |||||
| DATABASE Phix /cromwell_inputs/*/fastq_screen_reference/phix |
+ 2
- 0
inputSamplesFileExamples.tsv
查看文件
| oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R1.fastq.gz oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R2.fastq.gz oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam.bai oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Haplotyper/Fudan_DNA_LCL5_hc.vcf LCL5 | |||||
| oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL6_R1.fastq.gz oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL6_R2.fastq.gz oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Dedup/Fudan_DNA_LCL6.sorted.deduped.bam oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Dedup/Fudan_DNA_LCL6.sorted.deduped.bam.bai oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Haplotyper/Fudan_DNA_LCL6_hc.vcf LCL6 |
+ 23
- 0
inputs
查看文件
| { | |||||
| "{{ project_name }}.benchmarking_dir": "oss://chinese-quartet/quartet-result-data/NCTR_benchmarking_20181215/", | |||||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||||
| "{{ project_name }}.fastqc.disk_size": "150", | |||||
| "{{ project_name }}.benchmark.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||||
| "{{ project_name }}.fastqscreen.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||||
| "{{ project_name }}.fastqc.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||||
| "{{ project_name }}.benchmark.disk_size": "150", | |||||
| "{{ project_name }}.rtg.disk_size": "100", | |||||
| "{{ project_name }}.fastqc.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqc:v0.11.5", | |||||
| "{{ project_name }}.rtg.cluster_config": "OnDemand ecs.sn1ne.2xlarge img-ubuntu-vpc", | |||||
| "{{ project_name }}.benchmark.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/hap.py:latest", | |||||
| "{{ project_name }}.inputSamplesFile": "{{ inputSamplesFile }}", | |||||
| "{{ project_name }}.fastqscreen.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqscreen:0.12.0", | |||||
| "{{ project_name }}.screen_ref_dir": "oss://pgx-reference-data/fastq_screen_reference/", | |||||
| "{{ project_name }}.rtg.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||||
| "{{ project_name }}.fastq_screen_conf": "oss://chinese-quartet/quartet-storage-data/reference_data/fastq_screen.conf", | |||||
| "{{ project_name }}.bamqc.cluster_config": "OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc", | |||||
| "{{ project_name }}.fastqscreen.disk_size": "100", | |||||
| "{{ project_name }}.bamqc.disk_size": "500", | |||||
| "{{ project_name }}.bamqc.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/qualimap:2.0.0", | |||||
| "{{ project_name }}.ref_dir": "oss://chinese-quartet/quartet-storage-data/reference_data/" | |||||
| } |
二进制
pictures/.DS_Store
查看文件
二进制
pictures/Picture1.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|

|
|
| 宽度: 1122 | 高度: 622 | 大小: 240KB |
二进制
pictures/Screen Shot 2019-07-30 at 12.14.00 AM.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|

|
|
| 宽度: 1144 | 高度: 772 | 大小: 79KB |
二进制
pictures/Screen Shot 2019-07-31 at 12.40.56 AM.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|

|
|
| 宽度: 1322 | 高度: 1340 | 大小: 502KB |
二进制
pictures/density.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|
|
|
| 宽度: 1256 | 高度: 800 | 大小: 94KB |
+ 25
- 0
tasks/bamqc.wdl
查看文件
| task bamqc { | |||||
| File bam | |||||
| File bai | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| /opt/qualimap/qualimap bamqc -bam ${bam} -outformat PDF:HTML -nt $nt -outdir result --java-mem-size=32G | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster:cluster_config | |||||
| systemDisk:"cloud_ssd 40" | |||||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| Array[File] qualimap = glob("result/*") | |||||
| } | |||||
| } |
+ 51
- 0
tasks/benchmark.wdl
查看文件
| task benchmark { | |||||
| File gzvcf | |||||
| File gzvcf_index | |||||
| File benchmarking_dir | |||||
| File ref_dir | |||||
| String sample = basename(gzvcf,".vcf.gz") | |||||
| String sample_mark | |||||
| String fasta | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| export HGREF=/cromwell_inputs/*/reference_data/GRCh38.d1.vd1.fa | |||||
| if [ ${sample_mark} == "LCL5" ];then | |||||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL5.vcf.gz ${gzvcf} -f ${benchmarking_dir}/LCL5.bed.gz --threads $nt -o ${sample} | |||||
| elif [ ${sample_mark} == "LCL6" ]; then | |||||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL6.vcf.gz ${gzvcf} -f ${benchmarking_dir}/LCL6.bed.gz --threads $nt -o ${sample} | |||||
| elif [ ${sample_mark} == "LCL7" ]; then | |||||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL7.vcf.gz ${gzvcf} -f ${benchmarking_dir}/LCL6.bed.gz --threads $nt -o ${sample} | |||||
| elif [ ${sample_mark} == "LCL8" ]; then | |||||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL8.vcf.gz ${gzvcf} -f ${benchmarking_dir}/LCL6.bed.gz --threads $nt -o ${sample} | |||||
| else | |||||
| echo "only for quartet samples" | |||||
| fi | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster:cluster_config | |||||
| systemDisk:"cloud_ssd 40" | |||||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File gzip_vcf = "${sample}.vcf.gz" | |||||
| File gzip_vcf_index = "${sample}.vcf.gz.tbi" | |||||
| File roc_all_csv = "${sample}.roc.all.csv.gz" | |||||
| File roc_indel = "${sample}.roc.Locations.INDEL.csv.gz" | |||||
| File roc_indel_pass = "${sample}.roc.Locations.INDEL.PASS.csv.gz" | |||||
| File roc_snp = "${sample}.roc.Locations.SNP.csv.gz" | |||||
| File roc_snp_pass = "${sample}.roc.Locations.SNP.PASS.csv.gz" | |||||
| File summary = "${sample}.summary.csv" | |||||
| File extended = "${sample}.extended.csv" | |||||
| File metrics = "${sample}.metrics.json.gz" | |||||
| } | |||||
| } |
+ 28
- 0
tasks/fastqc.wdl
查看文件
| task fastqc { | |||||
| File read1 | |||||
| File read2 | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| fastqc -t $nt -o ./ ${read1} | |||||
| fastqc -t $nt -o ./ ${read2} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File read1_html = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||||
| File read1_zip = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||||
| File read2_html = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||||
| File read2_zip = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||||
| } | |||||
| } |
+ 34
- 0
tasks/fastqscreen.wdl
查看文件
| task fastq_screen { | |||||
| File read1 | |||||
| File read2 | |||||
| File screen_ref_dir | |||||
| File fastq_screen_conf | |||||
| String read1name = basename(read1,".fastq.gz") | |||||
| String read2name = basename(read2,".fastq.gz") | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read1} | |||||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read2} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File png1 = "${read1name}_screen.png" | |||||
| File txt1 = "${read1name}_screen.txt" | |||||
| File html1 = "${read1name}_screen.html" | |||||
| File png2 = "${read2name}_screen.png" | |||||
| File txt2 = "${read2name}_screen.txt" | |||||
| File html2 = "${read2name}_screen.html" | |||||
| } | |||||
| } |
+ 0
- 0
tasks/multiqc.wdl
查看文件
+ 23
- 0
tasks/zipindexVCF.wdl
查看文件
| task rtg { | |||||
| File vcf | |||||
| String sample = basename(vcf,".vcf") | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg bgzip ${vcf} -c > ${sample}.vcf.gz | |||||
| rtg index -f vcf ${sample}.vcf.gz | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File vcf_gz = "${sample}.vcf.gz" | |||||
| File vcf_index = "${sample}.vcf.gz.tbi" | |||||
| } | |||||
| } |
+ 55
- 0
workflow.wdl
查看文件
| import "./tasks/fastqc.wdl" as fastqc | |||||
| import "./tasks/fastqscreen.wdl" as fastqscreen | |||||
| import "./tasks/bamqc.wdl" as bamqc | |||||
| import "./tasks/zipindexVCF.wdl" as rtg | |||||
| import "./tasks/benchmark.wdl" as benchmark | |||||
| workflow {{ project_name }} { | |||||
| File inputSamplesFile | |||||
| Array[Array[File]] inputSamples = read_tsv(inputSamplesFile) | |||||
| File screen_ref_dir | |||||
| File fastq_screen_conf | |||||
| File benchmarking_dir | |||||
| File ref_dir | |||||
| String fasta | |||||
| scatter (sample in inputSamples){ | |||||
| call fastqc.fastqc as fastqc { | |||||
| input: | |||||
| read1=sample[0], | |||||
| read2=sample[1] | |||||
| } | |||||
| call fastqscreen.fastq_screen as fastqscreen { | |||||
| input: | |||||
| read1=sample[0], | |||||
| read2=sample[1], | |||||
| screen_ref_dir=screen_ref_dir, | |||||
| fastq_screen_conf=fastq_screen_conf | |||||
| } | |||||
| call bamqc.bamqc as bamqc { | |||||
| input: | |||||
| bam=sample[2], | |||||
| bai=sample[3] | |||||
| } | |||||
| call rtg.rtg as rtg { | |||||
| input: | |||||
| vcf=sample[4] | |||||
| } | |||||
| call benchmark.benchmark as benchmark { | |||||
| input: | |||||
| gzvcf=rtg.vcf_gz, | |||||
| gzvcf_index=rtg.vcf_index, | |||||
| benchmarking_dir=benchmarking_dir, | |||||
| ref_dir=ref_dir, | |||||
| sample_mark=sample[5], | |||||
| fasta=fasta | |||||
| } | |||||
| } | |||||
| } | |||||
正在加载...