

2 измененных файлов: 1 добавлений и 1 удалений
+ 1
- 1
README.md
Просмотреть файл
| @@ -12,7 +12,7 @@ | |||
|  | |||
|  | |||
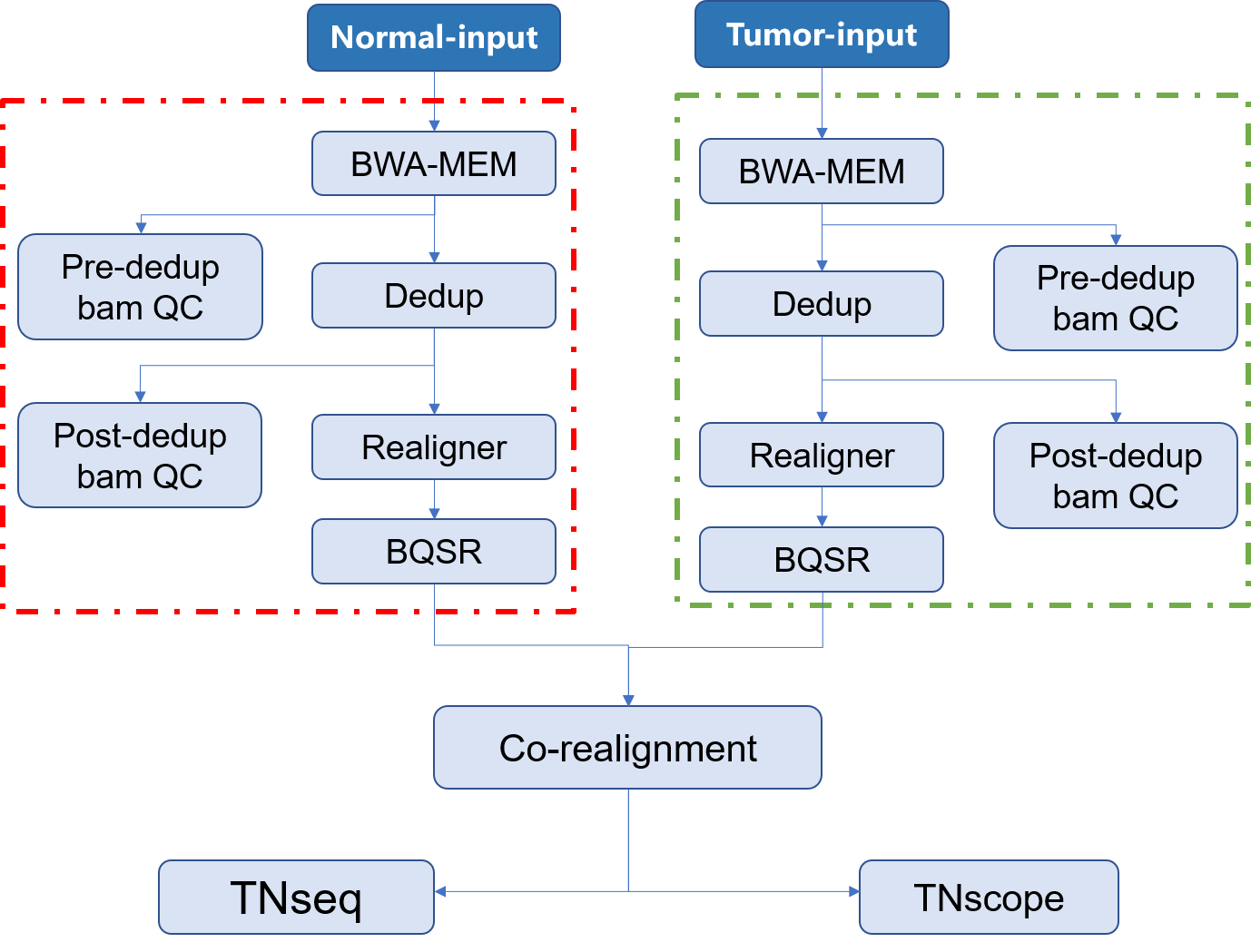
| 1. input:通常为二代目标区域测序所获得的fastq文件,通常包含 **Tumor** 和 **normal** 两种类型的数据;此外还应当包含有测序时使用的 `bed` 文件 | |||
| 2. Mapping:将测序所得的数据与参考基因组进行比对,找到每一条read在参考基因组上的位置,将结果信息储存在**bam**文件中,并对获得的 **bam** 文件进行质控 | |||
Двоичные данные
assets/somatic.png
Просмотреть файл
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1378 | Height: 1049 | Size: 97KB |
Загрузка…