

commit
1704d52cd1
16 ha cambiato i file con 473 aggiunte e 0 eliminazioni
+ 0
- 0
README.md
Vedi File
BIN
assets/%5CUsers%5Cthink%5CAppData%5CRoaming%5CTypora%5Ctypora-user-images%5C1547626023006.png
Vedi File
{kind=link}
| Before | After |
|---|---|
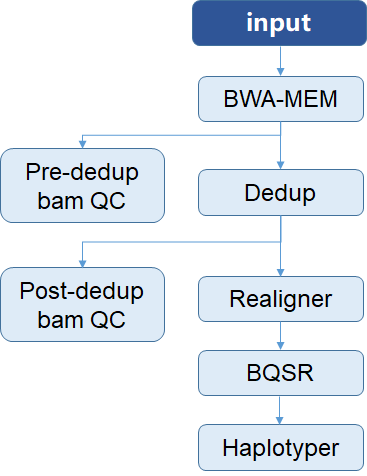
|
|
| Width: 367 | Height: 476 | Size: 18KB |
BIN
assets/1549784133993.png
Vedi File
{kind=link}
| Before | After |
|---|---|

|
|
| Width: 922 | Height: 464 | Size: 55KB |
BIN
assets/wgs-germline.png
Vedi File
{kind=link}
| Before | After |
|---|---|
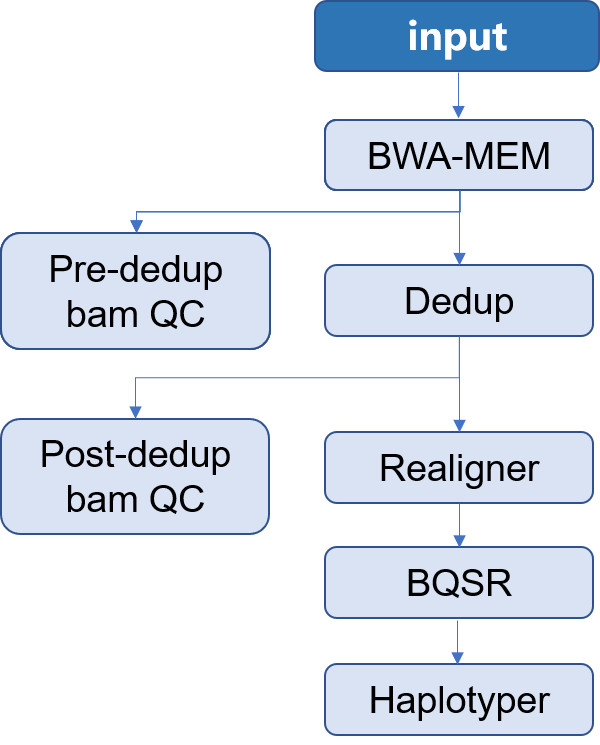
|
|
| Width: 600 | Height: 745 | Size: 21KB |
+ 14
- 0
inputs
Vedi File
| @@ -0,0 +1,14 @@ | |||
| { | |||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||
| "{{ project_name }}.ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "{{ project_name }}.dbsnp": "dbsnp_146.hg38.vcf", | |||
| "{{ project_name }}.SENTIEON_INSTALL_DIR": "/opt/sentieon-genomics", | |||
| "{{ project_name }}.dbmills_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "{{ project_name }}.db_mills": "Mills_and_1000G_gold_standard.indels.hg38.vcf", | |||
| "{{ project_name }}.cluster_config": "{{ cluster if cluster != '' else 'OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc' }}", | |||
| "{{ project_name }}.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/sentieon-genomics:v2018.08.01", | |||
| "{{ project_name }}.dbsnp_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "{{ project_name }}.sample": "{{ sample_name }}", | |||
| "{{ project_name }}.disk_size": 500 | |||
| } | |||
+ 48
- 0
tasks/BQSR.wdl
Vedi File
| @@ -0,0 +1,48 @@ | |||
| task BQSR { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File realigned_bam | |||
| File realigned_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} -q ${sample}_recal_data.table --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table.post --algo ReadWriter ${sample}.sorted.deduped.realigned.recaled.bam | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt --algo QualCal --plot --before ${sample}_recal_data.table --after ${sample}_recal_data.table.post ${sample}_recal_data.csv | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot QualCal -o ${sample}_bqsrreport.pdf ${sample}_recal_data.csv | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File recal_table = "${sample}_recal_data.table" | |||
| File recal_post = "${sample}_recal_data.table.post" | |||
| File recaled_bam = "${sample}.sorted.deduped.realigned.recaled.bam" | |||
| File recaled_bam_index = "${sample}.sorted.deduped.realigned.recaled.bam.bai" | |||
| File recal_csv = "${sample}_recal_data.csv" | |||
| File bqsrreport_pdf = "${sample}_bqsrreport.pdf" | |||
| } | |||
| } | |||
+ 40
- 0
tasks/Dedup.wdl
Vedi File
| @@ -0,0 +1,40 @@ | |||
| task Dedup { | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo LocusCollector --fun score_info ${sample}_score.txt | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo Dedup --rmdup --score_info ${sample}_score.txt --metrics ${sample}_dedup_metrics.txt ${sample}.sorted.deduped.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File score = "${sample}_score.txt" | |||
| File dedup_metrics = "${sample}_dedup_metrics.txt" | |||
| File Dedup_bam = "${sample}.sorted.deduped.bam" | |||
| File Dedup_bam_index = "${sample}.sorted.deduped.bam.bai" | |||
| } | |||
| } | |||
+ 38
- 0
tasks/Haplotyper.wdl
Vedi File
| @@ -0,0 +1,38 @@ | |||
| task Haplotyper { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File recaled_bam | |||
| File recaled_bam_index | |||
| String dbsnp | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper -d ${dbsnp_dir}/${dbsnp} ${sample}_hc.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf = "${sample}_hc.vcf" | |||
| File vcf_idx = "${sample}_hc.vcf.idx" | |||
| } | |||
| } | |||
+ 56
- 0
tasks/Metrics.wdl
Vedi File
| @@ -0,0 +1,56 @@ | |||
| task Metrics { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${sorted_bam} --algo MeanQualityByCycle ${sample}_mq_metrics.txt --algo QualDistribution ${sample}_qd_metrics.txt --algo GCBias --summary ${sample}_gc_summary.txt ${sample}_gc_metrics.txt --algo AlignmentStat ${sample}_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_is_metrics.txt --algo CoverageMetrics --omit_base_output ${sample}_coverage_metrics | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot metrics -o ${sample}_metrics_report.pdf gc=${sample}_gc_metrics.txt qd=${sample}_qd_metrics.txt mq=${sample}_mq_metrics.txt isize=${sample}_is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File qd_metrics = "${sample}_qd_metrics.txt" | |||
| File qd_metrics_pdf = "${sample}_qd_metrics.pdf" | |||
| File mq_metrics = "${sample}_mq_metrics.txt" | |||
| File mq_metrics_pdf = "${sample}_mq_metrics.pdf" | |||
| File is_metrics = "${sample}_is_metrics.txt" | |||
| File is_metrics_pdf = "${sample}_is_metrics.pdf" | |||
| File gc_summary = "${sample}_gc_summary.txt" | |||
| File gc_metrics = "${sample}_gc_metrics.txt" | |||
| File gc_metrics_pdf = "${sample}_gc_metrics.pdf" | |||
| File aln_metrics = "${sample}_aln_metrics.txt" | |||
| File coverage_metrics_sample_summary = "${sample}_coverage_metrics.sample_summary" | |||
| File coverage_metrics_sample_statistics = "${sample}_coverage_metrics.sample_statistics" | |||
| File coverage_metrics_sample_interval_statistics = "${sample}_coverage_metrics.sample_interval_statistics" | |||
| File coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File coverage_metrics_sample_cumulative_coverage_counts = "${sample}_coverage_metrics.sample_cumulative_coverage_counts" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/Realigner.wdl
Vedi File
| @@ -0,0 +1,41 @@ | |||
| task Realigner { | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String db_mills | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo Realigner -k ${dbmills_dir}/${db_mills} ${sample}.sorted.deduped.realigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File realigner_bam = "${sample}.sorted.deduped.realigned.bam" | |||
| File realigner_bam_index = "${sample}.sorted.deduped.realigned.bam.bai" | |||
| } | |||
| } | |||
+ 42
- 0
tasks/TNscope.wdl
Vedi File
| @@ -0,0 +1,42 @@ | |||
| task TNscope { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String tumor_name | |||
| String normal_name | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File corealigner_bam | |||
| File corealigner_bam_index | |||
| String dbsnp | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${corealigner_bam} --algo TNscope --tumor_sample ${tumor_name} --normal_sample ${normal_name} --dbsnp ${dbsnp_dir}/${dbsnp} ${sample}.TNscope.TN.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File TNscope_vcf= "${sample}.TNscope.TN.vcf" | |||
| File TNscope_vcf_index = "${sample}.TNscope.TN.vcf.idx" | |||
| } | |||
| } | |||
+ 43
- 0
tasks/TNseq.wdl
Vedi File
| @@ -0,0 +1,43 @@ | |||
| task TNseq { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String tumor_name | |||
| String normal_name | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File corealigner_bam | |||
| File corealigner_bam_index | |||
| String dbsnp | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${corealigner_bam} --algo TNhaplotyper --tumor_sample ${tumor_name} --normal_sample ${normal_name} --dbsnp ${dbsnp_dir}/${dbsnp} ${sample}.TNseq.TN.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File TNseq_vcf= "${sample}.TNseq.TN.vcf" | |||
| File TNseq_vcf_index = "${sample}.TNseq.TN.vcf.idx" | |||
| } | |||
| } | |||
+ 44
- 0
tasks/corealigner.wdl
Vedi File
| @@ -0,0 +1,44 @@ | |||
| task corealigner { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File tumor_recaled_bam | |||
| File tumor_recaled_bam_index | |||
| File normal_recaled_bam | |||
| File normal_recaled_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${tumor_recaled_bam} -i ${normal_recaled_bam} --algo Realigner -k ${db_mills} -k ${dbsnp} ${sample}_corealigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File corealigner_bam = "${sample}_corealigned.bam" | |||
| File corealigner_bam_index = "${sample}_corealigned.bam.bai" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/deduped_Metrics.wdl
Vedi File
| @@ -0,0 +1,37 @@ | |||
| task deduped_Metrics { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File deduped_coverage_metrics_sample_summary = "${sample}_deduped_coverage_metrics.sample_summary" | |||
| File deduped_coverage_metrics_sample_statistics = "${sample}_deduped_coverage_metrics.sample_statistics" | |||
| File deduped_coverage_metrics_sample_interval_statistics = "${sample}_deduped_coverage_metrics.sample_interval_statistics" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_counts = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_counts" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/mapping.wdl
Vedi File
| @@ -0,0 +1,34 @@ | |||
| task mapping { | |||
| File ref_dir | |||
| String fasta | |||
| File fastq_1 | |||
| File fastq_2 | |||
| String SENTIEON_INSTALL_DIR | |||
| String group | |||
| String sample | |||
| String pl | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i - | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File sorted_bam = "${sample}.sorted.bam" | |||
| File sorted_bam_index = "${sample}.sorted.bam.bai" | |||
| } | |||
| } | |||
+ 36
- 0
workflow.wdl
Vedi File
| @@ -0,0 +1,36 @@ | |||
| import "./tasks/Haplotyper.wdl" as Haplotyper | |||
| workflow {{ project_name }} { | |||
| File recaled_bam | |||
| File recaled_bam_index | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String docker | |||
| String fasta | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String db_mills | |||
| File dbsnp_dir | |||
| String dbsnp | |||
| String disk_size | |||
| String cluster_config | |||
| call Haplotyper.Haplotyper as Haplotyper { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=recaled_bam, | |||
| recaled_bam_index=recaled_bam_index, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| sample=sample, | |||
| docker=docker, | |||
| disk_size=disk_size, | |||
| cluster_config=cluster_config | |||
| } | |||
| } | |||
Loading…