
 LUYAO REN
5 年之前
LUYAO REN
5 年之前
當前提交
6d85d4ef49
共有 19 個文件被更改,包括 598 次插入 和 0 次删除
+ 94
- 0
fastq_screen.conf
查看文件
| @@ -0,0 +1,94 @@ | |||
| # This is an example configuration file for FastQ Screen | |||
| ############################ | |||
| ## Bowtie, Bowtie 2 or BWA # | |||
| ############################ | |||
| ## If the Bowtie, Bowtie 2 or BWA binary is not in your PATH, you can set | |||
| ## this value to tell the program where to find your chosen aligner. Uncomment | |||
| ## the relevant line below and set the appropriate location. Please note, | |||
| ## this path should INCLUDE the executable filename. | |||
| #BOWTIE /usr/local/bin/bowtie/bowtie | |||
| #BOWTIE2 /usr/local/bowtie2/bowtie2 | |||
| #BWA /usr/local/bwa/bwa | |||
| ############################################ | |||
| ## Bismark (for bisulfite sequencing only) # | |||
| ############################################ | |||
| ## If the Bismark binary is not in your PATH then you can set this value to | |||
| ## tell the program where to find it. Uncomment the line below and set the | |||
| ## appropriate location. Please note, this path should INCLUDE the executable | |||
| ## filename. | |||
| #BISMARK /usr/local/bin/bismark/bismark | |||
| ############ | |||
| ## Threads # | |||
| ############ | |||
| ## Genome aligners can be made to run across multiple CPU cores to speed up | |||
| ## searches. Set this value to the number of cores you want for mapping reads. | |||
| THREADS 32 | |||
| ############## | |||
| ## DATABASES # | |||
| ############## | |||
| ## This section enables you to configure multiple genomes databases (aligner index | |||
| ## files) to search against in your screen. For each genome you need to provide a | |||
| ## database name (which can't contain spaces) and the location of the aligner index | |||
| ## files. | |||
| ## | |||
| ## The path to the index files SHOULD INCLUDE THE BASENAME of the index, e.g: | |||
| ## /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 | |||
| ## Thus, the index files (Homo_sapiens.GRCh37.1.bt2, Homo_sapiens.GRCh37.2.bt2, etc.) | |||
| ## are found in a folder named 'GRCh37'. | |||
| ## | |||
| ## If, for example, the Bowtie, Bowtie2 and BWA indices of a given genome reside in | |||
| ## the SAME FOLDER, a SINLGE path may be provided to ALL the of indices. The index | |||
| ## used will be the one compatible with the chosen aligner (as specified using the | |||
| ## --aligner flag). | |||
| ## | |||
| ## The entries shown below are only suggested examples, you can add as many DATABASE | |||
| ## sections as required, and you can comment out or remove as many of the existing | |||
| ## entries as desired. We suggest including genomes and sequences that may be sources | |||
| ## of contamination either because they where run on your sequencer previously, or may | |||
| ## have contaminated your sample during the library preparation step. | |||
| ## | |||
| ## Human - sequences available from | |||
| ## ftp://ftp.ensembl.org/pub/current/fasta/homo_sapiens/dna/ | |||
| #DATABASE Human /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 | |||
| ## | |||
| ## Mouse - sequence available from | |||
| ## ftp://ftp.ensembl.org/pub/current/fasta/mus_musculus/dna/ | |||
| #DATABASE Mouse /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37 | |||
| ## | |||
| ## Ecoli- sequence available from EMBL accession U00096.2 | |||
| #DATABASE Ecoli /data/public/Genomes/Ecoli/Ecoli | |||
| ## | |||
| ## PhiX - sequence available from Refseq accession NC_001422.1 | |||
| #DATABASE PhiX /data/public/Genomes/PhiX/phi_plus_SNPs | |||
| ## | |||
| ## Adapters - sequence derived from the FastQC contaminats file found at: www.bioinformatics.babraham.ac.uk/projects/fastqc | |||
| #DATABASE Adapters /data/public/Genomes/Contaminants/Contaminants | |||
| ## | |||
| ## Vector - Sequence taken from the UniVec database | |||
| ## http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html | |||
| #DATABASE Vectors /data/public/Genomes/Vectors/Vectors | |||
| DATABASE Human /cromwell_root/tmp/fastq_screen_reference/genome | |||
| DATABASE Mouse /cromwell_root/tmp/fastq_screen_reference/mouse | |||
| DATABASE ERCC /cromwell_root/tmp/fastq_screen_reference/ERCC | |||
| DATABASE EColi /cromwell_root/tmp/fastq_screen_reference/ecoli | |||
| DATABASE Adapter /cromwell_root/tmp/fastq_screen_reference/adapters | |||
| DATABASE Vector /cromwell_root/tmp/fastq_screen_reference/vector | |||
| DATABASE rRNA /cromwell_root/tmp/fastq_screen_reference/rRNARef | |||
| DATABASE Virus /cromwell_root/tmp/fastq_screen_reference/viral | |||
| DATABASE Yeast /cromwell_root/tmp/fastq_screen_reference/GCF_000146045.2_R64_genomic_modify | |||
| DATABASE Mitoch /cromwell_root/tmp/fastq_screen_reference/Human_mitoch | |||
| DATABASE Phix /cromwell_root/tmp/fastq_screen_reference/phix | |||
+ 3
- 0
inputSamplesFileExamples.tsv
查看文件
| @@ -0,0 +1,3 @@ | |||
| #read1 #read2 #bam #bai #vcf #sample_mark #sample_name #_aln.metrics.txt #_dedup_metrics.txt #_is_metrics.txt #_deduped_coverage_metrics.sample_summary | |||
| oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R1.fastq.gz oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R2.fastq.gz oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam.bai oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Haplotyper/Fudan_DNA_LCL5_hc.vcf LCL5 | |||
| oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL6_R1.fastq.gz oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL6_R2.fastq.gz oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Dedup/Fudan_DNA_LCL6.sorted.deduped.bam oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Dedup/Fudan_DNA_LCL6.sorted.deduped.bam.bai oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/e85d0acb-f750-48b7-87e6-f28766dd16b9/call-Haplotyper/Fudan_DNA_LCL6_hc.vcf LCL6 | |||
+ 74
- 0
inputs
查看文件
| @@ -0,0 +1,74 @@ | |||
| { | |||
| "{{ project_name }}.benchmarking_dir": "oss://chinese-quartet/quartet-result-data/NCTR_benchmarking_20181215/", | |||
| "{{ project_name }}.vcfstat.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-hap:latest", | |||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||
| "{{ project_name }}.fastqc.disk_size": "150", | |||
| "{{ project_name }}.benchmark.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.mergeJI.disk_size": "100", | |||
| "{{ project_name }}.fastqscreen.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.fastqc.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.inputJIpiarsFile": "{{ inputJIpiarsFile }}", | |||
| "{{ project_name }}.benchmark.disk_size": "150", | |||
| "{{ project_name }}.vcfstat.disk_size": "100", | |||
| "{{ project_name }}.fastqc.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqc:v0.11.5", | |||
| "{{ project_name }}.mergeJI.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.benchmark.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-hap:latest", | |||
| "{{ project_name }}.inputSamplesFile": "{{ inputSamplesFile }}", | |||
| "{{ project_name }}.mergeJI.cluster_config": "OnDemand ecs.sn1ne.xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.fastqscreen.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqscreen:0.12.0", | |||
| "{{ project_name }}.mergeNum.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/gatk:v2019.01", | |||
| "{{ project_name }}.JI.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-hap:latest", | |||
| "{{ project_name }}.screen_ref_dir": "oss://pgx-reference-data/fastq_screen_reference/", | |||
| "{{ project_name }}.mergeNum.disk_size": "100", | |||
| "{{ project_name }}.JI.disk_size": "100", | |||
| "{{ project_name }}.fastq_screen_conf": "oss://pgx-reference-data/fastq_screen_reference/fastq_screen.conf", | |||
| "{{ project_name }}.multiqc.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.sdf": "oss://chinese-quartet/quartet-storage-data/reference_data/GRCh38.d1.vd1.sdf/", | |||
| "{{ project_name }}.multiqc.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/multiqc:v1.8", | |||
| "{{ project_name }}.mergeNum.cluster_config": "OnDemand ecs.sn1ne.xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.bamqc.cluster_config": "OnDemand ecs.sn1ne.8xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.vcfstat.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.JI.cluster_config": "OnDemand ecs.sn1ne.4xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.fastqscreen.disk_size": "100", | |||
| "{{ project_name }}.bamqc.disk_size": "500", | |||
| "{{ project_name }}.multiqc.disk_size": "100", | |||
| "{{ project_name }}.bamqc.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/qualimap:2.0.0", | |||
| "{{ project_name }}.ref_dir": "oss://chinese-quartet/quartet-storage-data/reference_data/" | |||
| } | |||
| { | |||
| "project_name.benchmarking_dir": "File", | |||
| "project_name.vcfstat.docker": "String", | |||
| "project_name.qualimap.docker": "String", | |||
| "project_name.qualimap.cluster_config": "String", | |||
| "project_name.fasta": "String", | |||
| "project_name.fastqc.disk_size": "String", | |||
| "project_name.mergeSentieon.docker": "String", | |||
| "project_name.benchmark.cluster_config": "String", | |||
| "project_name.sentieon.docker": "String", | |||
| "project_name.fastqscreen.cluster_config": "String", | |||
| "project_name.fastqc.cluster_config": "String", | |||
| "project_name.benchmark.disk_size": "String", | |||
| "project_name.vcfstat.disk_size": "String", | |||
| "project_name.fastqc.docker": "String", | |||
| "project_name.sentieon.cluster_config": "String", | |||
| "project_name.benchmark.docker": "String", | |||
| "project_name.inputSamplesFile": "File", | |||
| "project_name.fastqscreen.docker": "String", | |||
| "project_name.mergeNum.docker": "String", | |||
| "project_name.screen_ref_dir": "File", | |||
| "project_name.mergeNum.disk_size": "String", | |||
| "project_name.fastq_screen_conf": "File", | |||
| "project_name.multiqc.cluster_config": "String", | |||
| "project_name.qualimap.disk_size": "String", | |||
| "project_name.sdf": "File", | |||
| "project_name.multiqc.docker": "String", | |||
| "project_name.mergeNum.cluster_config": "String", | |||
| "project_name.mergeSentieon.disk_size": "String", | |||
| "project_name.sentieon.disk_size": "String", | |||
| "project_name.vcfstat.cluster_config": "String", | |||
| "project_name.mergeSentieon.cluster_config": "String", | |||
| "project_name.fastqscreen.disk_size": "String", | |||
| "project_name.multiqc.disk_size": "String", | |||
| "project_name.ref_dir": "File" | |||
| } | |||
二進制
pictures/.DS_Store
查看文件
二進制
pictures/Picture1.png
查看文件
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1122 | Height: 622 | Size: 240KB |
二進制
pictures/Screen Shot 2019-07-30 at 12.14.00 AM.png
查看文件
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1144 | Height: 772 | Size: 79KB |
二進制
pictures/Screen Shot 2019-07-31 at 12.40.56 AM.png
查看文件
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1322 | Height: 1340 | Size: 502KB |
二進制
pictures/density.png
查看文件
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 1256 | Height: 800 | Size: 94KB |
二進制
pictures/workflow2.png
查看文件
{kind=link}
| Before | After |
|---|---|
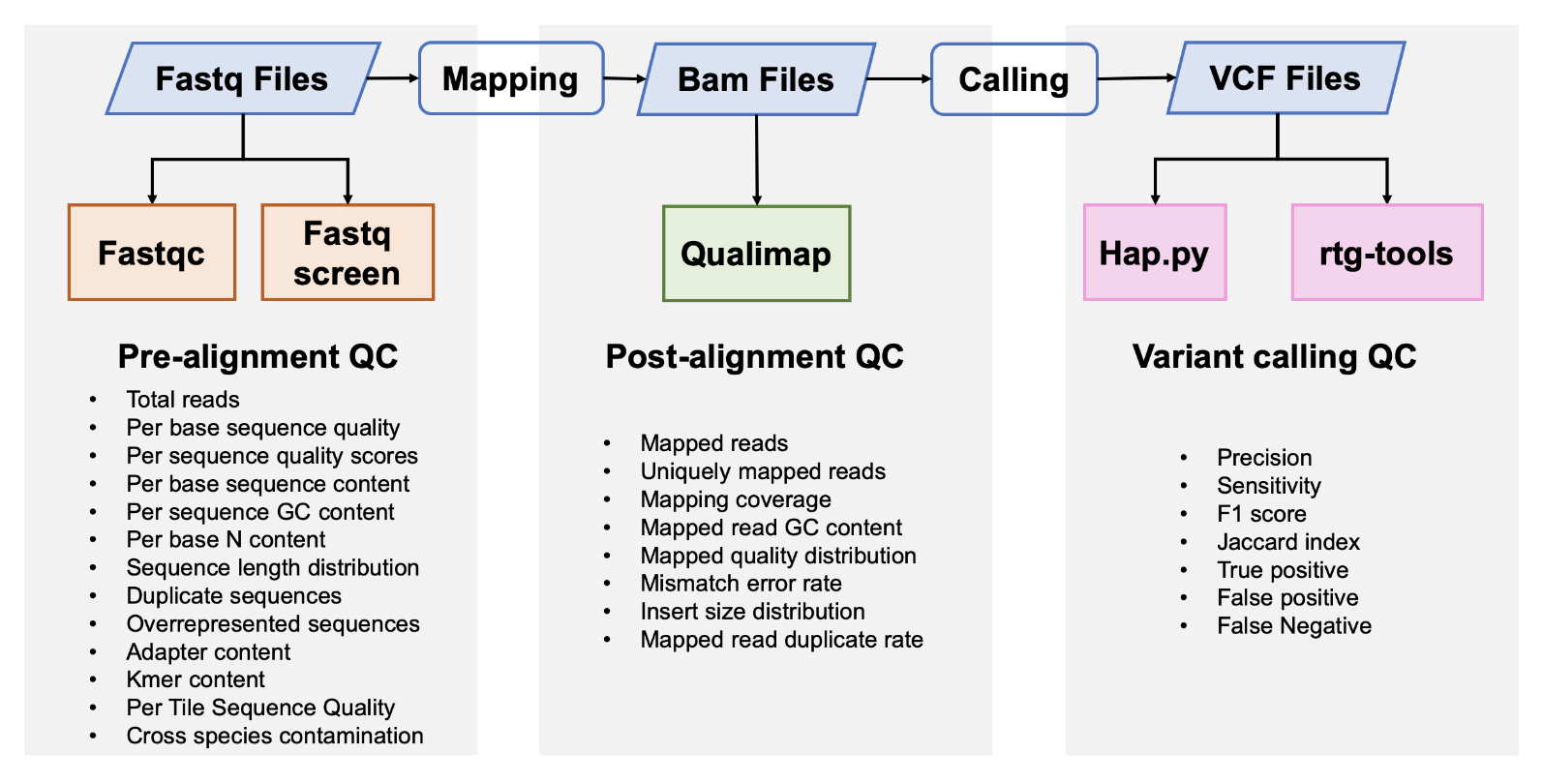
|
|
| Width: 1594 | Height: 810 | Size: 271KB |
+ 59
- 0
tasks/benchmark.wdl
查看文件
| @@ -0,0 +1,59 @@ | |||
| task benchmark { | |||
| File vcf | |||
| File benchmarking_dir | |||
| File ref_dir | |||
| String sample = basename(vcf,".vcf") | |||
| String sample_mark | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${ref_dir} /cromwell_root/tmp/ | |||
| export HGREF=/cromwell_root/tmp/reference_data/GRCh38.d1.vd1.fa | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip ${vcf} -c > ${sample}.rtg.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.rtg.vcf.gz | |||
| if [ ${sample_mark} == "LCL5" ];then | |||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL5.vcf.gz ${sample}.rtg.vcf.gz --threads $nt -o ${sample} | |||
| elif [ ${sample_mark} == "LCL6" ]; then | |||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL6.vcf.gz ${sample}.rtg.vcf.gz --threads $nt -o ${sample} | |||
| elif [ ${sample_mark} == "LCL7" ]; then | |||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL7.vcf.gz ${sample}.rtg.vcf.gz --threads $nt -o ${sample} | |||
| elif [ ${sample_mark} == "LCL8" ]; then | |||
| /opt/hap.py/bin/hap.py ${benchmarking_dir}/LCL8.vcf.gz ${sample}.rtg.vcf.gz --threads $nt -o ${sample} | |||
| else | |||
| echo "only for quartet samples" | |||
| fi | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File rtg_vcf = "${sample}.rtg.vcf.gz" | |||
| File rtg_vcf_index = "${sample}.rtg.vcf.gz.tbi" | |||
| File gzip_vcf = "${sample}.vcf.gz" | |||
| File gzip_vcf_index = "${sample}.vcf.gz.tbi" | |||
| File roc_all_csv = "${sample}.roc.all.csv.gz" | |||
| File roc_indel = "${sample}.roc.Locations.INDEL.csv.gz" | |||
| File roc_indel_pass = "${sample}.roc.Locations.INDEL.PASS.csv.gz" | |||
| File roc_snp = "${sample}.roc.Locations.SNP.csv.gz" | |||
| File roc_snp_pass = "${sample}.roc.Locations.SNP.PASS.csv.gz" | |||
| File summary = "${sample}.summary.csv" | |||
| File extended = "${sample}.extended.csv" | |||
| File metrics = "${sample}.metrics.json.gz" | |||
| } | |||
| } | |||
+ 28
- 0
tasks/fastqc.wdl
查看文件
| @@ -0,0 +1,28 @@ | |||
| task fastqc { | |||
| File read1 | |||
| File read2 | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| fastqc -t $nt -o ./ ${read1} | |||
| fastqc -t $nt -o ./ ${read2} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File read1_html = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||
| File read1_zip = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||
| File read2_html = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||
| File read2_zip = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||
| } | |||
| } | |||
+ 36
- 0
tasks/fastqscreen.wdl
查看文件
| @@ -0,0 +1,36 @@ | |||
| task fastq_screen { | |||
| File read1 | |||
| File read2 | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| String read1name = basename(read1,".fastq.gz") | |||
| String read2name = basename(read2,".fastq.gz") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${screen_ref_dir} /cromwell_root/tmp/ | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read1} | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read2} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File png1 = "${read1name}_screen.png" | |||
| File txt1 = "${read1name}_screen.txt" | |||
| File html1 = "${read1name}_screen.html" | |||
| File png2 = "${read2name}_screen.png" | |||
| File txt2 = "${read2name}_screen.txt" | |||
| File html2 = "${read2name}_screen.html" | |||
| } | |||
| } | |||
+ 26
- 0
tasks/mergeNum.wdl
查看文件
| @@ -0,0 +1,26 @@ | |||
| task mergeNum { | |||
| Array[File] vcfnumber | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| for i in ${sep=" " vcfnumber} | |||
| do | |||
| cat $i | cut -d':' -f2 | tr '\n' '\t' | sed s'/\t$/\n/g' >> vcfstats | |||
| done | |||
| sed '1i\File\tFailed Filters\tPassed Filters\tSNPs\tMNPs\tInsertions\tDeletions\tIndels\tSame as reference\tSNP Transitions/Transversions\tTotal Het/Hom ratio\tSNP Het/Hom ratio\tMNP Het/Hom ratio\tInsertion Het/Hom ratio\tDeletion Het/Hom ratio\tIndel Het/Hom ratio\tInsertion/Deletion ratio\tIndel/SNP+MNP ratio' vcfstats > vcfstats.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcfstat="vcfstats.txt" | |||
| } | |||
| } | |||
+ 40
- 0
tasks/mergeSentieon.wdl
查看文件
| @@ -0,0 +1,40 @@ | |||
| task mergeSentieon { | |||
| Array[File] aln_metrics_header | |||
| Array[File] aln_metrics_data | |||
| Array[File] dedup_metrics_header | |||
| Array[File] dedup_metrics_data | |||
| Array[File] is_metrics_header | |||
| Array[File] is_metrics_data | |||
| Array[File] deduped_coverage_header | |||
| Array[File] deduped_coverage_data | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| cat ${aln_metrics_header} | sed -n '1,1p' | cat - ${aln_metrics_data} > aln_metrics.txt | |||
| cat ${dedup_metrics_header} | sed -n '1,1p' | cat - ${dedup_metrics_data} > dedup_metrics.txt | |||
| cat ${is_metrics_header} | sed -n '1,1p' | cat - ${is_metrics_data} > is_metrics.txt | |||
| cat ${deduped_coverage_header} | sed -n '1,1p' | cat - ${deduped_coverage_data} > deduped_coverage.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File aln_metrics_merge = "aln_metrics.txt" | |||
| File dedup_metrics_merge = "dedup_metrics.txt" | |||
| File is_metrics_merge = "is_metrics.txt" | |||
| File deduped_coverage_merge = "deduped_coverage.txt" | |||
| } | |||
| } | |||
+ 48
- 0
tasks/multiqc.wdl
查看文件
| @@ -0,0 +1,48 @@ | |||
| task multiqc { | |||
| Array[File] read1_zip | |||
| Array[File] read2_zip | |||
| Array[File] txt1 | |||
| Array[File] txt2 | |||
| Array[File] zip | |||
| Array[File] summary | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| mkdir -p /cromwell_root/tmp/fastqc | |||
| mkdir -p /cromwell_root/tmp/fastqscreen | |||
| mkdir -p /cromwell_root/tmp/bamqc | |||
| mkdir -p /cromwell_root/tmp/benchmark | |||
| cp ${sep=" " read1_zip} ${sep=" " read2_zip} /cromwell_root/tmp/fastqc | |||
| cp ${sep=" " txt1} ${sep=" " txt2} /cromwell_root/tmp/fastqscreen | |||
| cp ${sep=" " summary} /cromwell_root/tmp/benchmark | |||
| for i in ${sep=" " zip} | |||
| do | |||
| tar -zxvf $i -C /cromwell_root/tmp/bamqc | |||
| done | |||
| multiqc /cromwell_root/tmp/ | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File multiqc_html = "multiqc_report.html" | |||
| Array[File] multiqc_txt = glob("multiqc_data/*") | |||
| } | |||
| } | |||
+ 27
- 0
tasks/qualimap.wdl
查看文件
| @@ -0,0 +1,27 @@ | |||
| task qualimap { | |||
| File bam | |||
| File bai | |||
| String bamname = basename(bam,".bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| /opt/qualimap/qualimap bamqc -bam ${bam} -outformat PDF:HTML -nt $nt -outdir ${bamname} --java-mem-size=32G | |||
| tar -zcvf ${bamname}_qualimap.zip ${bamname} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File zip = "${bamname}_qualimap.zip" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/sentieon.wdl
查看文件
| @@ -0,0 +1,41 @@ | |||
| task sentieon { | |||
| File aln_metrics | |||
| File dedup_metrics | |||
| File is_metrics | |||
| File deduped_coverage | |||
| String sample_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| cat ${aln_metrics} | sed -n '2,2p' > aln_metrics.header | |||
| cat ${aln_metrics} | sed -n '2,2p' > ${sample_name}.aln_metrics | |||
| cat ${dedup_metrics} | sed -n '2,2p' > dedup_metrics.header | |||
| cat ${dedup_metrics} | sed -n '3,3p' > ${sample_name}.dedup_metrics | |||
| cat ${is_metrics} | sed -n '2,2p' > is_metrics.header | |||
| cat ${is_metrics} | sed -n '3,3p' > ${sample_name}.is_metrics | |||
| cat ${deduped_coverage} | sed -n '1,1p' > deduped_coverage.header | |||
| cat ${deduped_coverage} | sed -n '2,2p' > ${sample_name}.deduped_coverage | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File aln_metrics_header = "aln_metrics.header" | |||
| File aln_metrics_data = "${sample_name}.aln_metrics" | |||
| File dedup_metrics_header = "dedup_metrics.header" | |||
| File dedup_metrics_data = "${sample_name}.dedup_metrics" | |||
| File is_metrics_header = "is_metrics.header" | |||
| File is_metrics_data = "${sample_name}.is_metrics" | |||
| File deduped_coverage_header = "deduped_coverage.header" | |||
| File deduped_coverage_data = "${sample_name}.deduped_coverage" | |||
| } | |||
| } | |||
+ 25
- 0
tasks/vcfstat.wdl
查看文件
| @@ -0,0 +1,25 @@ | |||
| task vcfstat { | |||
| File rtg_vcf | |||
| File rtg_vcf_index | |||
| String sample_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg vcfstats ${rtg_vcf} > ${sample_name}.stats.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcfnumber="${sample_name}.stats.txt" | |||
| } | |||
| } | |||
+ 97
- 0
workflow.wdl
查看文件
| @@ -0,0 +1,97 @@ | |||
| import "./tasks/fastqc.wdl" as fastqc | |||
| import "./tasks/fastqscreen.wdl" as fastqscreen | |||
| import "./tasks/qualimap.wdl" as qualimap | |||
| import "./tasks/benchmark.wdl" as benchmark | |||
| import "./tasks/vcfstat.wdl" as vcfstat | |||
| import "./tasks/sentieon.wdl" as sentieon | |||
| import "./tasks/multiqc.wdl" as multiqc | |||
| import "./tasks/mergeNum.wdl" as mergeNum | |||
| import "./tasks/mergeSentieon.wdl" as mergeSentieon | |||
| workflow project_name { | |||
| File inputSamplesFile | |||
| Array[Array[File]] inputSamples = read_tsv(inputSamplesFile) | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| File benchmarking_dir | |||
| File ref_dir | |||
| String fasta | |||
| File sdf | |||
| scatter (sample in inputSamples) { | |||
| call fastqc.fastqc as fastqc { | |||
| input: | |||
| read1=sample[0], | |||
| read2=sample[1] | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen { | |||
| input: | |||
| read1=sample[0], | |||
| read2=sample[1], | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf | |||
| } | |||
| call qualimap.qualimap as qualimap { | |||
| input: | |||
| bam=sample[2], | |||
| bai=sample[3] | |||
| } | |||
| call benchmark.benchmark as benchmark { | |||
| input: | |||
| vcf=sample[4], | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| sample_mark=sample[5], | |||
| fasta=fasta | |||
| } | |||
| call vcfstat.vcfstat as vcfstat { | |||
| input: | |||
| rtg_vcf=benchmark.rtg_vcf, | |||
| rtg_vcf_index=benchmark.rtg_vcf_index, | |||
| sample_name=sample[6] | |||
| } | |||
| call sentieon.sentieon as sentieon { | |||
| input: | |||
| aln_metrics=sample[7], | |||
| dedup_metrics=sample[8], | |||
| is_metrics=sample[9], | |||
| deduped_coverage=sample[10], | |||
| sample_name=sample[6] | |||
| } | |||
| } | |||
| call multiqc.multiqc as multiqc { | |||
| input: | |||
| read1_zip=fastqc.read1_zip, | |||
| read2_zip=fastqc.read2_zip, | |||
| txt1=fastqscreen.txt1, | |||
| txt2=fastqscreen.txt2, | |||
| zip=qualimap.zip, | |||
| summary=benchmark.summary | |||
| } | |||
| call mergeNum.mergeNum as mergeNum { | |||
| input: | |||
| vcfnumber=vcfstat.vcfnumber | |||
| } | |||
| call mergeSentieon.mergeSentieon as mergeSentieon { | |||
| input: | |||
| aln_metrics_header=sentieon.aln_metrics_header, | |||
| aln_metrics_data=sentieon.aln_metrics_data, | |||
| dedup_metrics_header=sentieon.dedup_metrics_header, | |||
| dedup_metrics_data=sentieon.dedup_metrics_data, | |||
| is_metrics_header=sentieon.is_metrics_header, | |||
| is_metrics_data=sentieon.is_metrics_data, | |||
| deduped_coverage_header=sentieon.deduped_coverage_header, | |||
| deduped_coverage_data=sentieon.deduped_coverage_data | |||
| } | |||
| } | |||
Loading…