
 LUYAO REN
438941c49b
read me
LUYAO REN
438941c49b
read me
|
4 anni fa | |
|---|---|---|
| codescripts | 4 anni fa | |
| pictures | 4 anni fa | |
| tasks | 4 anni fa | |
| README.md | 4 anni fa | |
| choppy_samples_example.csv | 4 anni fa | |
| defaults | 4 anni fa | |
| inputSamples.Example.txt | 4 anni fa | |
| inputs | 4 anni fa | |
| workflow.wdl | 4 anni fa | |
README.md
Quality control of germline variants calling results using a Chinese Quartet family
Author: Run Luyao
E-mail:18110700050@fudan.edu.cn
Git: http://47.103.223.233/renluyao/quartet_dna_quality_control_wgs_big_pipeline
Last Updates: 2021/7/5
Install
open-choppy-env
choppy install renluyao/quartet_dna_quality_control_big_pipeline
Introduction of Chinese Quartet DNA reference materials
With the rapid development of sequencing technology and the dramatic decrease of sequencing costs, DNA sequencing has been widely used in scientific research, diagnosis of and treatment selection for human diseases. However, due to the lack of effective quality assessment and control of the high-throughput omics data generation and analysis processes, variants calling results are seriously inconsistent among different technical replicates, batches, laboratories, sequencing platforms, and analysis pipelines, resulting in irreproducible scientific results and conclusions, huge waste of resources, and even endangering the life and health of patients. Therefore, reference materials for quality control of the whole process from omics data generation to data analysis are urgently needed.
We first established genomic DNA reference materials from four immortalized B-lymphoblastoid cell lines of a Chinese Quartet family including parents and monozygotic twin daughters to make performance assessment of germline variants calling results. To establish small variant benchmark calls and regions, we generated whole-genome sequencing data in nine batches, with depth ranging from 30x to 60x, by employing PCR-free and PCR libraries on four popular short-read sequencing platforms (Illumina HiSeq XTen, Illumina NovaSeq, MGISEQ-2000, and DNBSEQ-T7) with three replicates at each batch, resulting in 108 libraries in total and 27 libraries for each Quartet DNA reference material. Then, we selected variants concordant in multiple call sets and in Mendelian consistency within Quartet family members as small variant benchmark calls, resulting in 4.2 million high-confidence variants (SNV and Indel) and 2.66 G high confidence genomic region, covering 87.8% of the human reference genome (GRCh38, chr1-22 and X). Two orthogonal technologies were used for verifying the high-confidence variants. The consistency rate with PMRA (Axiom Precision Medicine Research Array) was 99.6%, and 95.9% of high-confidence variants were validated by 10X Genomics whole-genome sequencing data. Genetic built-in truth of the Quartet family design is another kind of “truth” within the four Quartet samples. Apart from comparison with benchmark calls in the benchmark regions to identify false-positive and false-negative variants, pedigree information among the Quartet DNA reference materials, i.e., reproducibility rate of variants between the twins and Mendelian concordance rate among family members, are complementary approaches to comprehensively estimate genome-wide variants calling performance. Finally, we developed a whole-genome sequencing data quality assessment pipeline and demonstrated its utilities with two examples of using the Quartet reference materials and datasets to evaluate data generation performance in three sequencing labs and different data analysis pipelines.
Softwares and parameters
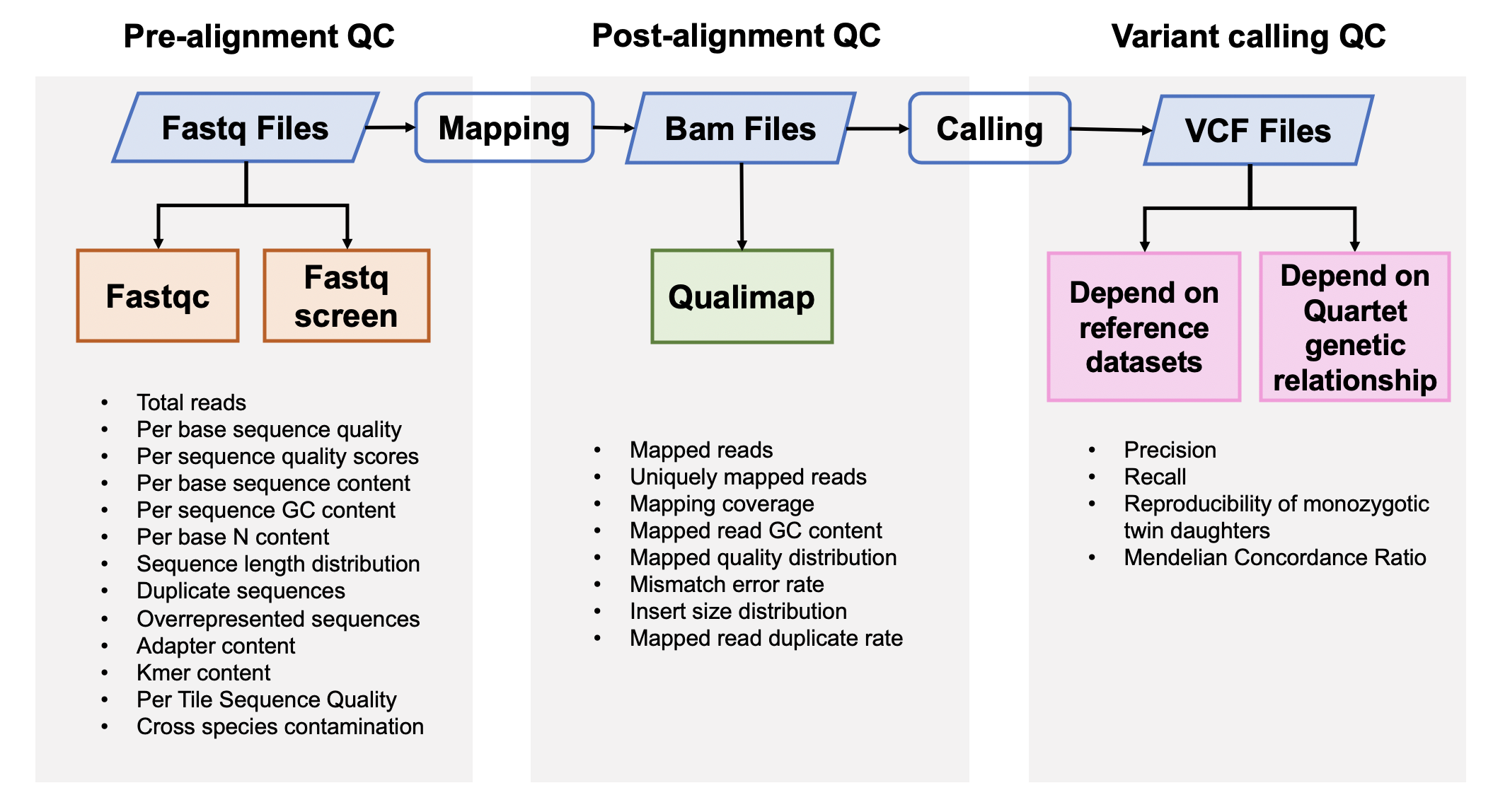
1. Pre-alignment QC
Fastqc v0.11.5
FastQC is used to investigate the quality of fastq files
fastqc -t <threads> -o <output_directory> <fastq_file>
Fastq Screen 0.12.0
Fastq Screen is used to inspect whether the library were contaminated. For example, we expected 99% reads aligned to human genome, 10% reads aligned to mouse genome, which is partly homologous to human genome. If too many reads are aligned to E.Coli or Yeast, libraries or cell lines are probably comtminated.
fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file>
2. Post-alignment QC
Qualimap 2.0.0
Qualimap is used to check the quality od bam files
qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G
3. Variants Calling QC
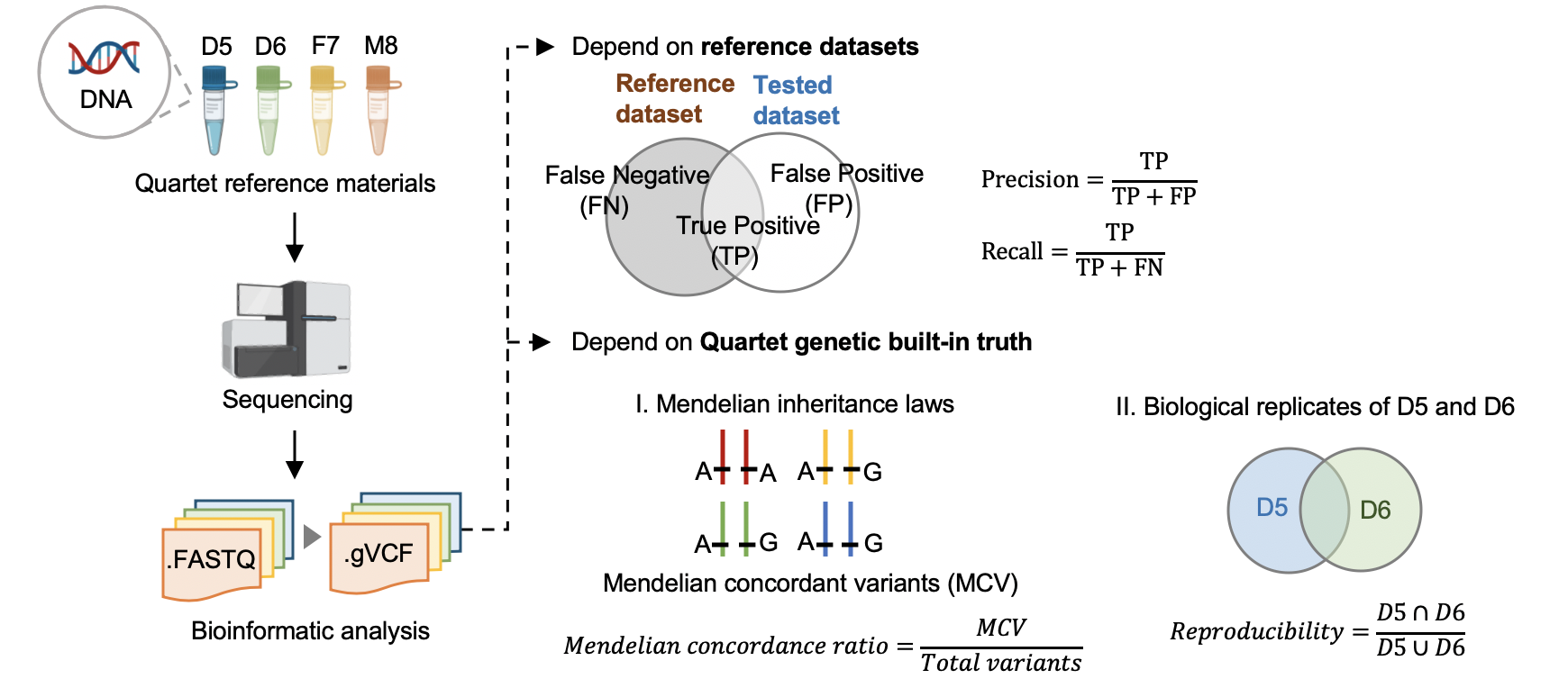
3.1 Performance assessment based on reference datasets
Hap.py v0.3.9
hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename>
3.2 Performance assessment based on Quartet genetic built-in truth
Mendelian Concordance Rate (vbt v1.1)
We splited the Quartet family to two trios (F7, M8, D5 and F7, M8, D6) and then do the Mendelian analysis. A Quartet Mendelian concordant variant is the same between the twins (D5 and D6) , and follow the Mendelian concordant between parents (F7 and M8). Mendelian concordance rate is the Mendelian concordance variant divided by total detected variants in a Quartet family.
vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads>
Input files
choppy samples renluyao/quartet_dna_quality_control_wgs_big_pipeline-latest --output samples
Samples CSV file
1. Start from Fastq files
sample_id,project,fastq_1_D5,fastq_2_D5,fastq_1_D6,fastq_2_D6,fastq_1_F7,fastq_2_F7,fastq_1_M8,fastq_2_M8
# sample_id in choppy system
# project name
# oss path of D5 fastq read1 file
# oss path of D5 fastq read2 file
# oss path of D6 fastq read1 file
# oss path of D6 fastq read2 file
# oss path of F7 fastq read1 file
# oss path of F7 fastq read2 file
# oss path of M8 fastq read1 file
# oss path of M8 fastq read2 file
2. Start from VCF files
sample_id,project,vcf_D5,vcf_D6,vcf_F7,vcf_M8
# sample_id in choppy system
# project name
# oss path of D5 VCF file
# oss path of D6 VCF file
# oss path of F7 VCF file
# oss path of M8 VCF file
Output Files
1. extract_tables.wdl/extract_tables_vcf.wdl
(FASTQ) Pre-alignment QC: pre_alignment.txt
(FASTQ) Post-alignment QC: post_alignment.txt
(FASTQ/VCF) Variants calling QC: variants.calling.qc.txt
2. quartet_mendelian.wdl
(FASTQ/VCF) Mendelian concordance rate: mendelian.txt
结果展示与解读
1. pre_alignment.txt
| Column name | Description |
|---|---|
| Sample | 样本名,R1结尾为read1,R2结尾为read2 |
| %Dup | % Duplicate reads |
| %GC | Average % GC content |
| Total Sequences (million) | Total sequences |
| %Human | 比对到人类基因组的比例 |
| %EColi | 比对到大肠杆菌基因组的比例 |
| %Adapter | 比对到接头序列的比例 |
| %Vector | 比对到载体基因组的比例 |
| %rRNA | 比对到rRNA序列的比例 |
| %Virus | 比对到病毒基因组的比例 |
| %Yeast | 比对到酵母基因组的比例 |
| %Mitoch | 比对到线粒体序列的比例 |
| %No hits | 没有比对到以上基因组的比例 |
2. post_alignment.txt
| Column name | Description |
|---|---|
| Sample | 样本名 |
| %Mapping | % mapped reads |
| %Mismatch Rate | Mapping error rate |
| Mendelian Insert Size | Median insert size(bp) |
| %Q20 | % bases >Q20 |
| %Q30 | % bases >Q30 |
| Mean Coverage | Mean deduped coverage |
| Median Coverage | Median deduped coverage |
| PCT_1X | Fraction of genome with at least 1x coverage |
| PCT_5X | Fraction of genome with at least 5x coverage |
| PCT_10X | Fraction of genome with at least 10x coverage |
| PCT_30X | Fraction of genome with at least 30x coverage |
3. variants.calling.qc.txt
| Column name | Description |
|---|---|
| Sample | 样本名 |
| SNV number | 检测到SNV的数目 |
| INDEL number | 检测到INDEL的数目 |
| SNV query | 在高置信基因组区域中的SNV数目 |
| INDEL query | 在高置信基因组区域中INDEL数目 |
| SNV TP | 真阳性SNV |
| INDEL TP | 真阳性INDEL |
| SNV FP | 假阳性SNV |
| INDEL FP | 假阳性INDEL |
| SNV FN | 假阴性SNV |
| INDEL FN | 假阴性INDEL |
| SNV precision | SNV与标准集比较的precision |
| INDEL precision | INDEL的与标准集比较的precision |
| SNV recall | SNV与标准集比较的recall |
| INDEL recall | INDEL的与标准集比较的recall |
| SNV F1 | SNV与标准集比较的F1-score |
| INDEL F1 | INDEL与标准集比较的F1-score |
4 mendelian.txt
| Column name | Description |
|---|---|
| Family | 家庭名字,我们目前的设计是4个Quartet样本,每个三个技术重复,family_1是指rep1的4个样本组成的家庭单位,以此类推。 |
| Total_Variants | 四个Quartet样本一共能检测到的变异位点数目 |
| Mendelian_Concordant_Variants | 符合孟德尔规律的变异位点数目 |
| Mendelian_Concordance_Quartet | 符合孟德尔遗传的比例 |