
22KB
WGS-germline Small Variants Quality Control Pipeline
Author: Run Luyao
E-mail:18110700050@fudan.edu.cn
Git: http://choppy.3steps.cn/renluyao/WGS_germline_datapotal.git
Last Updates: 2020/07/05
安装指南
# 激活choppy环境
source activate choppy
# 安装app
choppy install renluyao/WGS_germline_datapotal
App概述——中华家系1号标准物质介绍
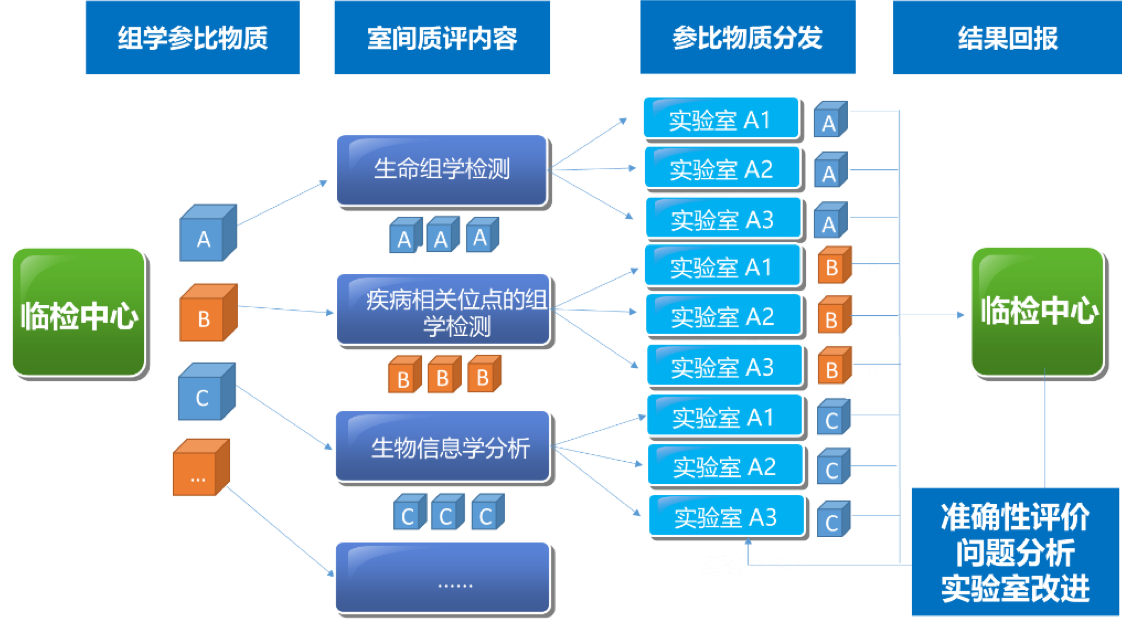
建立高通量全基因组测序的生物计量和质量控制关键技术体系,是保障测序数据跨技术平台、跨实验室可比较、相关研究结果可重复、数据可共享的重要关键共性技术。建立国家基因组标准物质和基准数据集,突破基因组学的生物计量技术,是将测序技术转化成临床应用的重要环节与必经之路,目前国际上尚属空白。中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制了人源中华家系1号基因组标准物质(Quartet,一套4个样本,编号分别为LCL5,LCL6,LCL7,LCL8,其中LCL5和LCL6为同卵双胞胎女儿,LCL7为父亲,LCL8为母亲),以及相应的全基因组测序序列基准数据集(“量值”),为衡量基因序列检测准确与否提供一把“标尺”,成为保障基因测序数据可靠性的国家基准。人源中华家系1号基因组标准物质来源于泰州队列同卵双生双胞胎家庭,从遗传结构上体现了我国南北交界的人群结构特征,同时家系的设计也为“量值”的确定提供了遗传学依据。
中华家系1号DNA标准物质的Small Variants标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。此外,我们还可根据中华家系1号的生物遗传关系计算同卵双胞胎检测突变的一致性和符合四口之家遗传规律的一致率估计测序错误的比例,评估数据产生和分析的质量好坏。
该Quality_control APP用于全基因组测序(whole-genome sequencing,WGS)数据的质量评估,包括原始数据质控、比对数据质控和突变检出数据质控。
流程与参数
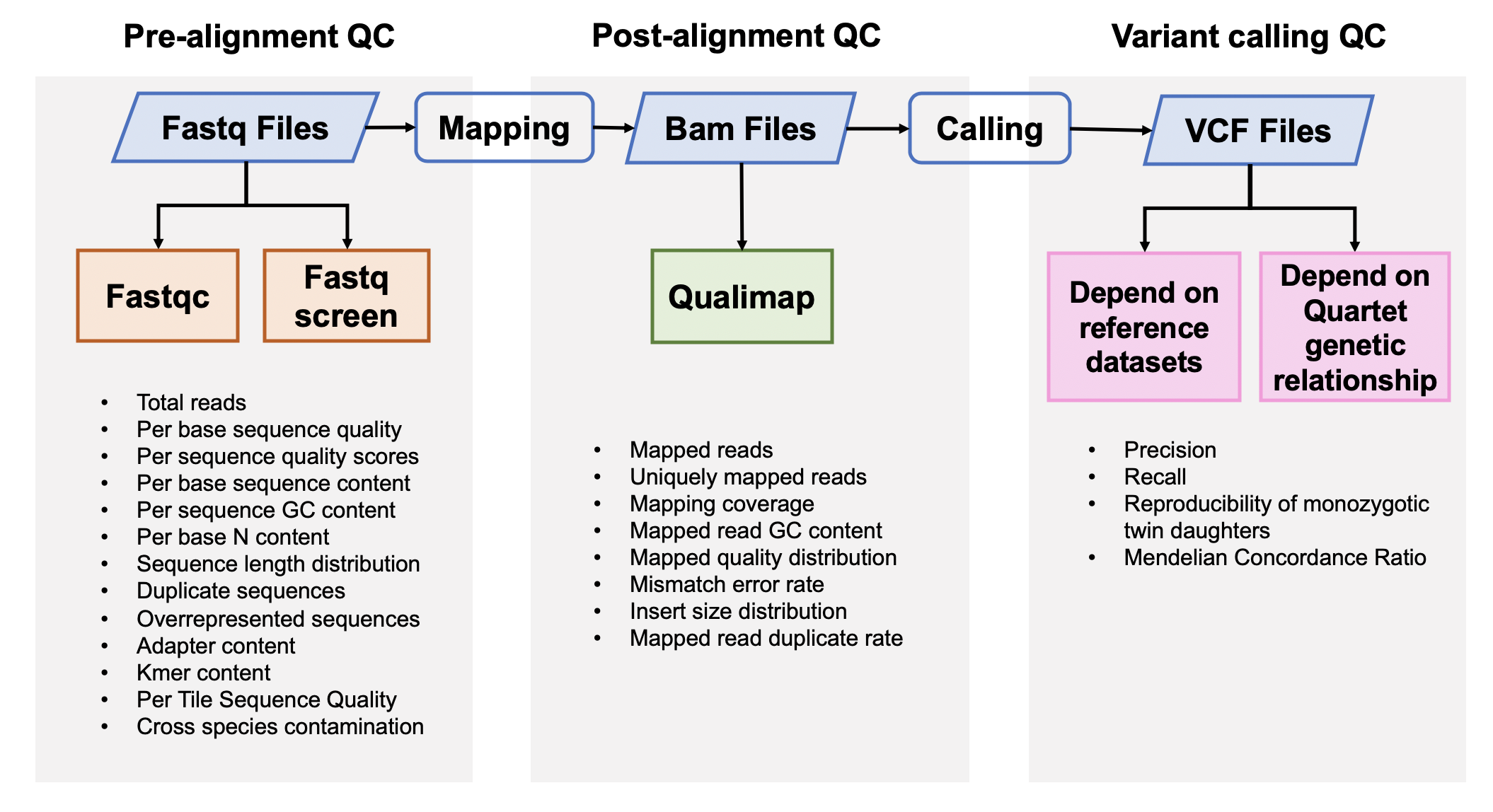
1. 原始数据质量控制
Fastqc v0.11.5
FastQC是一个常用的测序原始数据的质控软件,主要包括12个模块,具体请参考Fastqc模块详情。
fastqc -t <threads> -o <output_directory> <fastq_file>
Fastq Screen 0.12.0
Fastq Screen是检测测序原始数据中是否引⼊入其他物种,或是接头引物等污染,⽐比如,如果测序样本 是⼈人类,我们期望99%以上的reads匹配到⼈人类基因组,10%左右的reads匹配到与⼈人类基因组同源性 较⾼高的⼩小⿏鼠上。如果有过多的reads匹配到Ecoli或者Yeast,要考虑是否在培养细胞的时候细胞系被污 染,或者建库时⽂文库被污染。
fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file>
--conf conifg 文件主要输入了多个物种的fasta文件地址,可根据自己自己的需求下载其他物种的fasta文件加入分析
--top一般不需要对整个fastq文件进行检索,取前100000行
2. 比对后数据质量控制
Qualimap 2.0.0
Qualimap是一个比对指控软件,包含Picard的MarkDuplicates的结果和sentieon中metrics的质控结果。
qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G
3. 突变检出数据质量控制
突变质量控制的流程如下
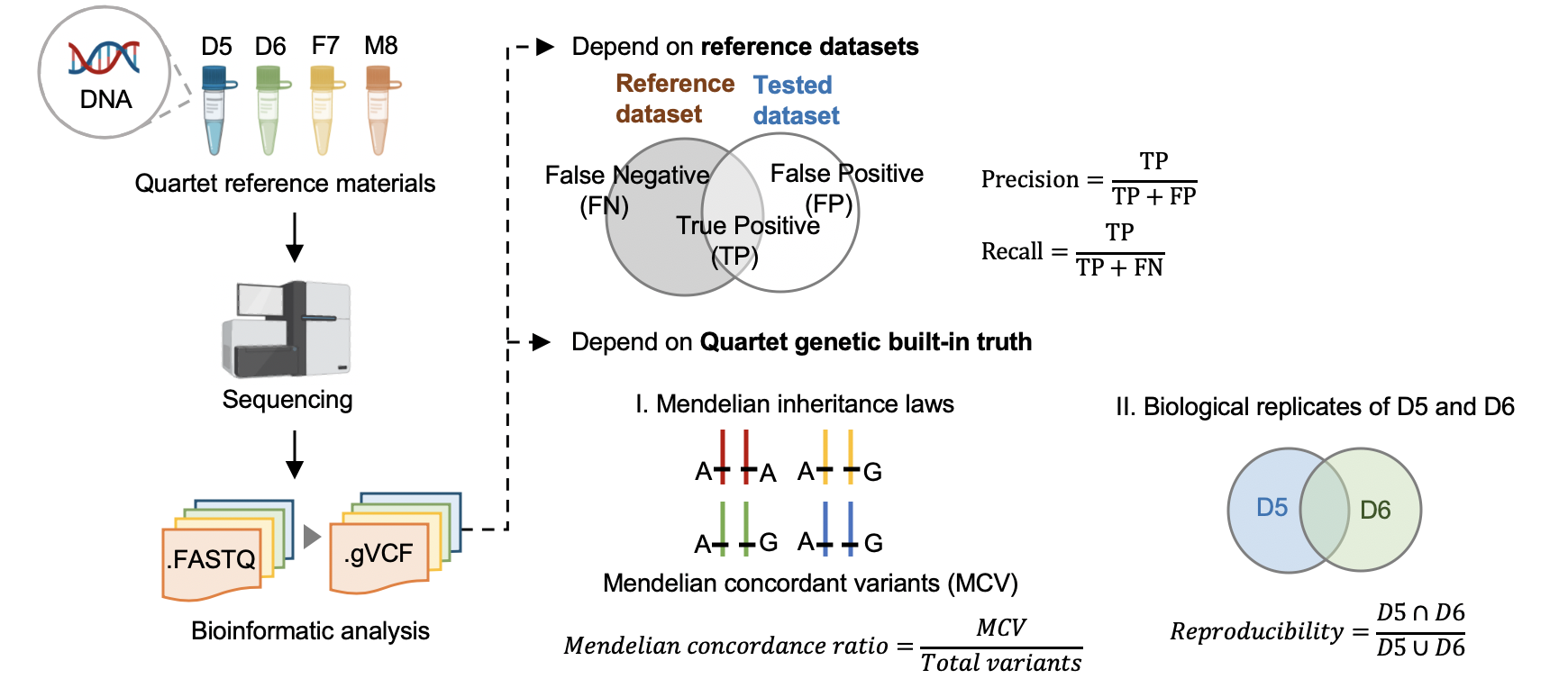
3.1 根据标准数据集的数据质量控制
Hap.py v0.3.9
hap.py是将被检测vcf结果与benchmarking对比,计算precision和recall的软件,它考虑了vcf中突变表示形式的多样性,进行了归一化。
hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename>
3.2 根据Quartet四口之家遗传规律的质量控制
Reproducibility (in-house python script)
标准数据集是根据我们整合多个平台方法,过滤不可重复检测、不符合孟德尔遗传规律的假阳性的突变。它可以评估数据产生和分析方法的相对好坏,但是具有一定的局限性,因为它排除掉了很多难测的基因组区域。我们可以通过比较同卵双胞胎突变检测的一致性对全基因组范围进行评估。
Mendelian Concordance Ratio (vbt v1.1)
我们首先将四口之家拆分成两个三口之家进行孟德尔遗传的分析。当一个突变符合姐妹一致,且与父母符合孟德尔遗传规律,则认为是符合Quartet四口之家的孟德尔遗传规律。孟德尔符合率是指四个标准检测出的所有突变中满足孟德尔遗传规律的比例。
vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads>
App输入文件
choppy samples WGS_germline_datapotal-latest --output samples
Samples文件的输入包括
inputSamplesFile
#read1 #read2 #sample_name
read1 是阿里云上fastq read1的地址
read2 是阿里云上fastq read2的地址
sample_name是指样本的命名
所有上传的文件应有规范的命名
Quartet_DNA_SequenceTech_SequenceMachine_SequenceSite_Sample_Replicate_Date.R1/R2.fastq.gz
SequenceTech是指测序平台,如ILM、BGI等
SequenceMachine是指测序仪器,如XTen、Nova、Hiseq(Illumina)、SEQ500、SEQ1000(BGI)等
SequenceSite是指测序单位的英文缩写
Sample是指LCL5、LCL6、LCL7、LCL8
Replicate是指技术重复,从1开始依次增加
Date是指数据获得日期,格式为20200710
后缀一定是R1/R2.fastq.gz,不可以随意更改,R1/R2不可以写成r1/r2,fastq.gz不可以写成fq.gz
各个缩写规范请见 https://fudan-pgx.yuque.com/docs/share/5baa851b-da97-47b9-b6c4-78f2b60595ab?# 《数据命名规范》
project
这个项目的名称,可以写自己可以识别的字符串,只能写英文和数字,不可以写中文
Quartet样本的组合问题
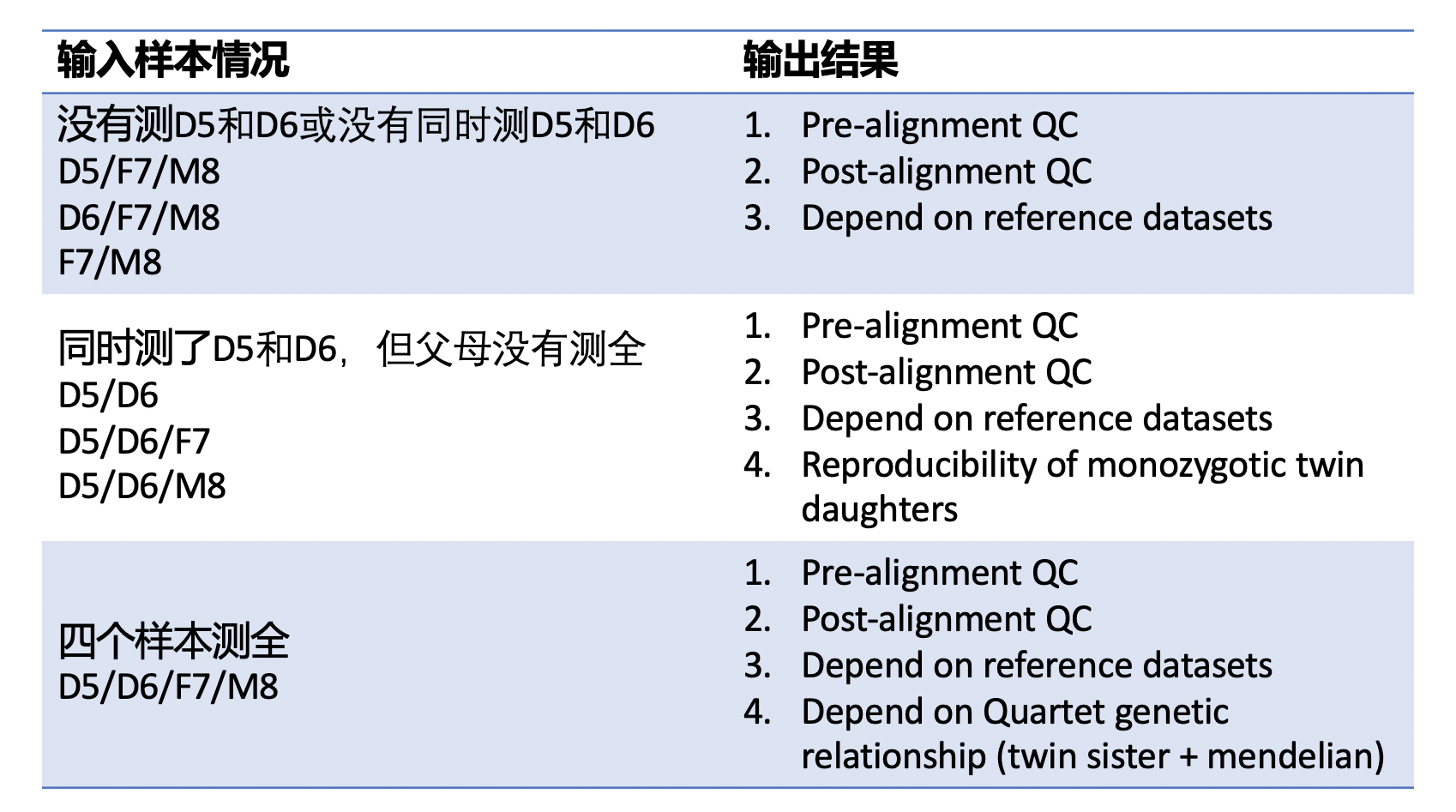
1. 没有测LCL5和LCL6,或者没有同时测LCL5和LCL6
只给出原始数据质控、比对数据质控、与标准集的比较
2. 包含LCL5和LCL6同卵双胞胎的数据,但是父母的数据不全
只给出原始数据质控、比对数据质控、与标准集的比较、同卵双胞胎一致性
3. 四个quartet样本都测了
给出所有结果原始数据质控、比对数据质控、与标准集的比较、同卵双胞胎一致性,符合孟德尔遗传比例
注意:本app假设每个批次测的技术重复都一样,如batch 1测了LCL5、LCL6、LCL7和LCL8,batch 2 和batch 3也都测了这四个样本。本app不解决特别复杂的问题,例如batch1测了LCL5,LCL6,batch2测了LCL7和LCL8,本app只能给出原始数据质控、比对数据质控、与标准集的比较,不会把多个批次的数据合并计算孟德尔符合率和姐妹一致性。
App输出文件
本计算会产生大量的中间结果,这里说明最后整合好的结果文件,上述的三种情况得到的结果文件如下。
1. 没有测LCL5和LCL6,或者没有同时测LCL5和LCL6
1.1 原始数据质量控制
1.2 比对后数据质量控制
输出目录
extract_multiqc
输出结果文件
qualimap.final.result.txt
1.3 突变检出数据质量控制
与标准集进行比较
输出目录
extract_multiqc
输出结果文件
benchmark.final.result.txt
2. 包含LCL5和LCL6同卵双胞胎的数据,但是父母的数据不全
2.1 原始数据质量控制
输出目录
extract_multiqc
输出结果文件
fastqc.final.result.txt
fastqscreen.final.result.txt
2.2 比对后数据质量控制
输出目录
extract_multiqc
输出结果文件
qualimap.final.result.txt
2.3 突变检出数据质量控制
2.3.1 与标准集进行比较
输出目录
extract_multiqc
输出结果文件
benchmark.final.result.txt
2.3.2 姐妹一致性
输出目录
D5_D6
输出结果文件
${project}.sister.txt
3. 四个quartet样本都测了
3.1 原始数据质量控制
输出目录
extract_multiqc
输出结果文件
fastqc.final.result.txt
fastqscreen.final.result.txt
3.2 比对后数据质量控制
输出目录
extract_multiqc
输出结果文件
qualimap.final.result.txt
3.3 突变检出数据质量控制
3.3.1 与标准集进行比较
输出目录
extract_multiqc
输出结果文件
benchmark.final.result.txt
3.3.2 通过Quartet家系设计
输出目录
quartet_mendelian
输出结果文件
${project}.mendelian.txt
结果展示与解读
1. 原始数据质量控制
原始数据质量控制主要通过考察测序数据的基本特征判断数据质量的好坏,比如数据量是否达到要求、reads的重复率是否过多、碱基质量、ATGC四种碱基的分布、GC含量、接头序列含量以及是否有其他物种的污染等等。
FastQC和FastqScreen是两个常用的原始数据质量控制软件
1.1 fastqc.final.result.txt
| 列名 | 说明 |
|---|---|
| Sample | 样本名,R1结尾为read1,R2结尾为read2 |
| FastQC_mqc-generalstats-fastqc-percent_duplicates | % Duplicate reads |
| FastQC_mqc-generalstats-fastqc-percent_gc | Average % GC content |
| FastQC_mqc-generalstats-fastqc-avg_sequence_length | Reads长度 |
| FastQC_mqc-generalstats-fastqc-percent_fails | 12个Fastqc模块中失败的比例 |
| FastQC_mqc-generalstats-fastqc-total_sequences | Total sequences |
| per_base_sequence_quality | pass/warn/fail;Quality就是Fred值,-10*log10(p),p为测错的概率。所以一条reads某位置出错概率为0.01时,其quality就是20。横轴代表位置,纵轴quality。红色表示中位数,黄色是25%-75%区间,触须是10%-90%区间,蓝线是平均数。若任一位置的下四分位数低于10或中位数低于25,报”WARN”;若任一位置的下四分位数低于5或中位数低于20,报”FAIL”。 |
| per_tile_sequence_quality | pass/warn/fail;每个tile的测序情况,横轴是测序序列第1个碱基到第101个碱基,纵轴是tail的Index编号,这个图主要是为了防止,在测序过程中,某些tail受到不可控因素的影响而出现测序质量偏低,蓝色代表测序质量很高,暖色代表测序质量不高,如果某些tail出现暖色,可以在后续分析中把该tail测序的结果全部都去除。 |
| per_sequence_quality_scores | pass/warn/fail;每条reads的quality的均值的分布,横轴为quality,纵轴是reads数目。当出现上图的情况时,我们就会知道有一部分reads具有比较差的质量。当峰值小于27(错误率0.2%)时报”WARN”,当峰值小于20(错误率1%)时报”FAIL”。 |
| per_base_sequence_content | pass/warn/fail;对所有reads的每一个位置,统计ATCG四种碱基(正常情况)的分布:横轴为位置,纵轴为百分比。 正常情况下四种碱基的出现频率应该是接近的,而且没有位置差异。因此好的样本中四条线应该平行且接近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。当任一位置的A/T比例与G/C比例相差超过10%,报”WARN”;当任一位置的A/T比例与G/C比例相差超过20%,报”FAIL”。 |
| per_sequence_gc_content | pass/warn/fail;统计reads的平均GC含量的分布。红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。偏离理论分布的reads超过15%时,报”WARN”;偏离理论分布的reads超过30%时,报”FAIL”。 |
| per_base_n_content | pass/warn/fail;当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”。对所有reads的每个位置,统计N的比率:正常情况下N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。当任意位置的N的比例超过5%,报”WARN”;当任意位置的N的比例超过20%,报”FAIL”。 |
| sequence_length_distribution | pass/warn/fail;reads长度的分布。当reads长度不一致时报”WARN”;当有长度为0的read时报“FAIL”。 |
| sequence_duplication_levels | pass/warn/fail;统计序列完全一样的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在(如建库过程中的PCR duplication)。横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。 如果原始数据很大(事实往往如此),做这样的统计将非常慢,所以fastqc中用fq数据的前200,000条reads统计其在全部数据中的重复情况。重复数目大于等于10的reads被合并统计,这也是为什么我们看到上图的最右侧略有上扬。大于75bp的reads只取50bp(不知道怎么选的)进行比较。但由于reads越长越不容易完全相同(由测序错误导致),所以其重复程度仍有可能被低估。当非unique的reads占总数的比例大于20%时,报”WARN”;当非unique的reads占总数的比例大于50%时,报”FAIL“。 |
| overrepresented_sequences | pass/warn/fail;如果有某个序列大量出现,就叫做over-represented。fastqc的标准是占全部reads的0.1%以上。和上面的duplicate analysis一样,为了计算方便,只取了fq数据的前200,000条reads进行统计,所以有可能over-represented reads不在里面。而且大于75bp的reads也是只取50bp。如果命令行中加入了-c contaminant file,出现的over-represented sequence会从contaminant_file里面找匹配的hit(至少20bp且最多一个mismatch),可以给我们一些线索。当发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“。 |
| adapter_content | pass/warn/fail;序列中两端adapter的情况;如果有adapter序列没有去除干净的情况,在后续分析的时候需要先使用cutadapt软件进行去接头,也可以用 trimmomatic来去除接头。 |
| kmer_content | pass/warn/fail;如果某k个bp的短序列在reads中大量出现,其频率高于统计期望的话,fastqc将其记为over-represented k-mer。默认的k = 5,可以用-k --kmers选项来调节,范围是2-10。出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer被认为是over-represented。fastqc除了列出所有over-represented k-mers,还会把前6个的per base distribution画出来。当有出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer时,报”WARN“;当有出现频率在某位置上10倍于期望的k-mer时报”FAIL”。 |
1.2 fastqscreen.final.result.txt
| 列名 | 说明 |
|---|---|
| Sample | 样本名,R1结尾为read1,R2结尾为read2 |
| Human percentage | 比对到人类基因组的比例 |
| ERCC percentage | 比对到External RNA Controls Consortium基因组的比例 |
| EColi percentage | 比对到大肠杆菌基因组的比例 |
| Adapter percentage | 比对到接头序列的比例 |
| Vector percentage | 比对到载体基因组的比例 |
| rRNA percentage | 比对到rRNA序列的比例 |
| Virus percentage | 比对到病毒基因组的比例 |
| Yeast percentage | 比对到酵母基因组的比例 |
| Mitoch percentage | 比对到线粒体序列的比例 |
| Phix percentage | 比对到Phix基因组的比例。PhiX对照品v3是一款可靠、连接接头的文库,适合用作Illumina测序运行的对照品。 |
| No hits percentage | 没有比对到以上基因组的比例 |
2. 比对后数据质量控制
qualimap.final.result.txt
| 列名 | 说明 |
|---|---|
| Sample | 样本名 |
| QualiMap_mqc-generalstats-qualimap-avg_gc | Mean GC content |
| QualiMap_mqc-generalstats-qualimap-median_insert_size | Median insert size(bp) |
| QualiMap_mqc-generalstats-qualimap-1_x_pc | Fraction of genome with at least 1x coverage |
| QualiMap_mqc-generalstats-qualimap-5_x_pc | Fraction of genome with at least 5x coverage |
| QualiMap_mqc-generalstats-qualimap-10_x_pc | Fraction of genome with at least 10x coverage |
| QualiMap_mqc-generalstats-qualimap-30_x_pc | Fraction of genome with at least 30x coverage |
| QualiMap_mqc-generalstats-qualimap-50_x_pc | Fraction of genome with at least 50x coverage |
| QualiMap_mqc-generalstats-qualimap-median_coverage | Median coverage |
| QualiMap_mqc-generalstats-qualimap-percentage_aligned | % mapped reads |
| QualiMap_mqc-generalstats-qualimap-mapped_reads | Number of mapped reads |
| QualiMap_mqc-generalstats-qualimap-total_reads | Total reads |
| QualiMap_mqc-generalstats-qualimap-general_error_rate | Mapping error rate |
3. 突变检出数据质量控制
3.1 与标准集对比 benchmark.final.result.txt
| 列名 | 说明 |
|---|---|
| Sample | 样本名 |
| Precision | 查准率 |
| Recall | 查全率 |
3.2 姐妹一致性 ${project}.sister.txt (样本没有没有测全,但是同时测了LCL5和LCL6)
| 列名 | 说明 |
|---|---|
| Family | 家庭名字,我们目前的设计是4个Quartet样本,每个三个技术重复,family_1是指rep1的4个样本组成的家庭单位,以此类推。 |
| Reproducibility_D5_D6 | Quartet-D5和Quartet-D6的一致性 |
3.3 Quartet家系关系评估 ${project}.mendelian.txt
| 列名 | 说明 |
|---|---|
| Family | 家庭名字,我们目前的设计是4个Quartet样本,每个三个技术重复,family_1是指rep1的4个样本组成的家庭单位,以此类推。 |
| Reproducibility_D5_D6 | Quartet-D5和Quartet-D6的一致性 |
| Mendelian_Concordance_Quartet | 符合孟德尔遗传的比例 |