
8.3KB
WGS-germline Small Variants Quality Control Pipeline(Start from VCF files)
Author: Run Luyao
E-mail:18110700050@fudan.edu.cn
Git: http://choppy.3steps.cn/renluyao/quartet-wgs-germline-data-analysis-qc.git
Last Updates: 2021/04/14
安装指南
# 激活choppy环境
open-choppy-env
# 安装app
choppy install renluyao/quartet-wgs-germline-data-analysis-qc
App概述——中华家系1号标准物质介绍
建立高通量全基因组测序的生物计量和质量控制关键技术体系,是保障测序数据跨技术平台、跨实验室可比较、相关研究结果可重复、数据可共享的重要关键共性技术。建立国家基因组标准物质和基准数据集,突破基因组学的生物计量技术,是将测序技术转化成临床应用的重要环节与必经之路,目前国际上尚属空白。中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制了人源中华家系1号基因组标准物质(Quartet,一套4个样本,编号分别为LCL5,LCL6,LCL7,LCL8,其中LCL5和LCL6为同卵双胞胎女儿,LCL7为父亲,LCL8为母亲),以及相应的全基因组测序序列基准数据集(“量值”),为衡量基因序列检测准确与否提供一把“标尺”,成为保障基因测序数据可靠性的国家基准。人源中华家系1号基因组标准物质来源于泰州队列同卵双生双胞胎家庭,从遗传结构上体现了我国南北交界的人群结构特征,同时家系的设计也为“量值”的确定提供了遗传学依据。
中华家系1号DNA标准物质的Small Variants标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。此外,我们还可根据中华家系1号的生物遗传关系计算同卵双胞胎检测突变的一致性和符合四口之家遗传规律的一致率估计测序错误的比例,评估数据产生和分析的质量好坏。
2021年5-7月在临检中心的领导下组织全国临床和科研实验室全外显子测序室间质量评价预研https://www.nccl.org.cn/showEqaPtDetail?id=1514
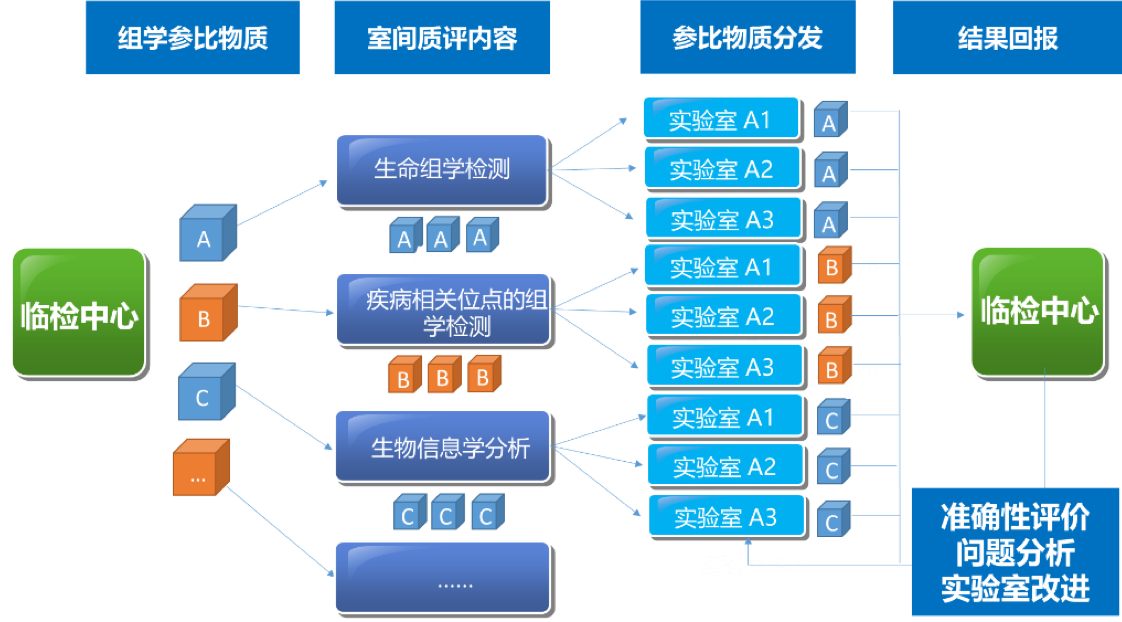
该Quality_control APP用于全基因组测序(whole-exome sequencing,WES)数据的质量评估,包括原始数据质控、比对数据质控和突变检出数据质控。
流程与参数
突变质量控制的流程如下
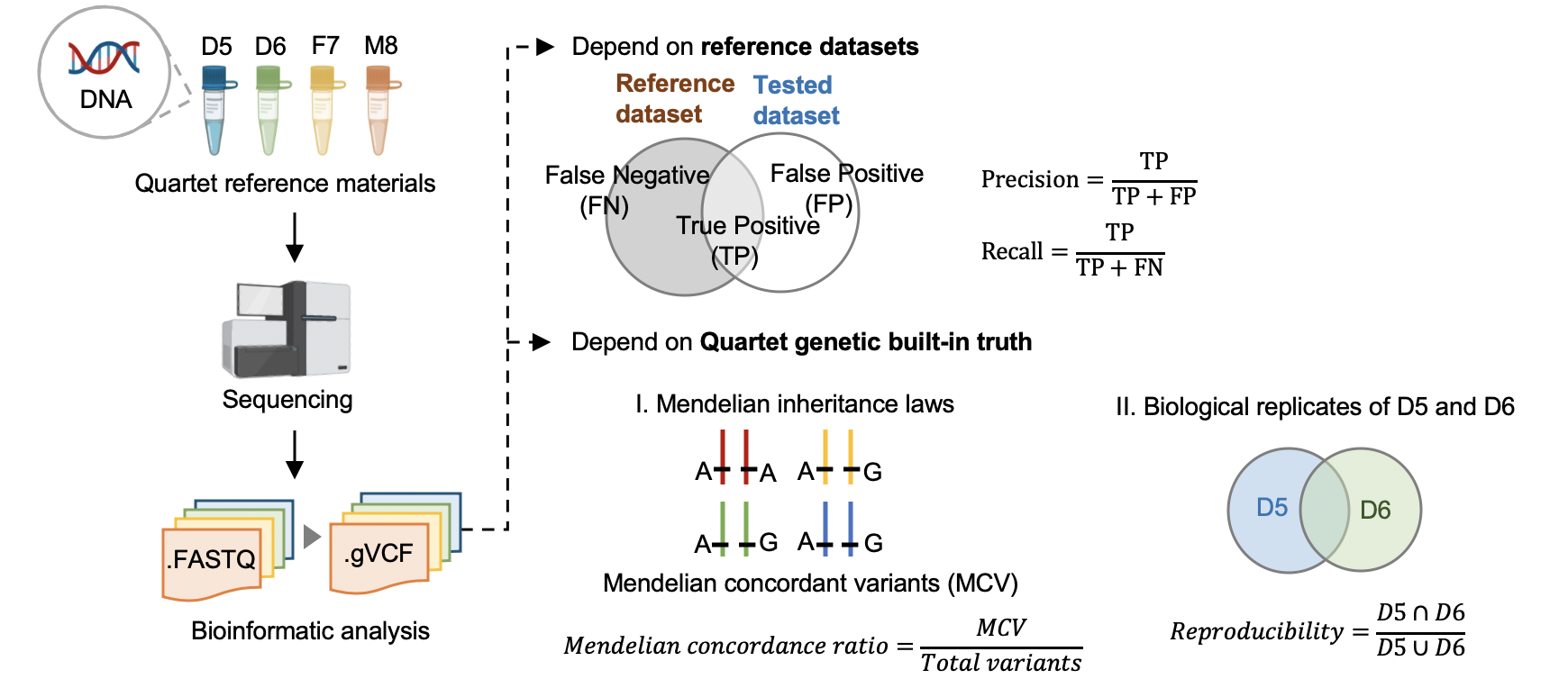
1 根据标准数据集的数据质量控制
Hap.py v0.3.9
hap.py是将被检测vcf结果与benchmarking对比,计算precision和recall的软件,它考虑了vcf中突变表示形式的多样性,进行了归一化。
hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename>
2 根据Quartet四口之家遗传规律的质量控制
Reproducibility (in-house python script)
标准数据集是根据我们整合多个平台方法,过滤不可重复检测、不符合孟德尔遗传规律的假阳性的突变。它可以评估数据产生和分析方法的相对好坏,但是具有一定的局限性,因为它排除掉了很多难测的基因组区域。我们可以通过比较同卵双胞胎突变检测的一致性对全基因组范围进行评估。
Mendelian Concordance Ratio (vbt v1.1)
我们首先将四口之家拆分成两个三口之家进行孟德尔遗传的分析。当一个突变符合姐妹一致,且与父母符合孟德尔遗传规律,则认为是符合Quartet四口之家的孟德尔遗传规律。孟德尔符合率是指四个标准检测出的所有突变中满足孟德尔遗传规律的比例。
vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads>
App输入文件
choppy samples renluyao/quartet-wes-germline-data-analysis-qc-latest --output samples
Samples文件的输入包括
1. inputSamplesFile,该文件的上传至阿里云,samples文件中填写该文件的阿里云地址
请查看示例 vcf_qc_inputsamplesfiles.tsv
#LCL5_1 #LCL6_1 #LCL7_1 #LCL8_1 1
#LCL5_2 #LCL6_2 #LCL7_2 #LCL8_2 2
#LCL5_3 #LCL6_3 #LCL7_3 #LCL8_3 3
目前本流程只能分析12个一组的样本,每一行是四个Quartet样本的VCF阿里云地址,第5列是数字序号,用于区分replicate
2. project
这个项目的名称,可以写自己可以识别的字符串,只能写英文和数字,不可以写中文
samples文件的示例请查看choppy_samples_example.csv
App输出文件
本计算会产生大量的中间结果,这里说明最后整合好的结果文件。在一个tasks输出最终的结果:
quartet_mendelian.wdl
突变检出指控 variants.calling.qc.txt
如果用户输入4个一组完整的家系样本则可以得到每个家庭单位的precision和recall的平均值,用于报告第一页的展示:
reference_datasets_aver-std.txt
基于Quartet家系的质控 ${project}.mendelian.txt
Quartet家系结果的平均值和SD值,用于报告第一页的展示
quartet_indel_aver-std.txt
quartet_snv_aver-std.txt
结果展示与解读
1 与标准数据集进行比较
具体信息variants.calling.qc.txt
| 列名 | 说明 |
|---|---|
| Sample | 样本名 |
| SNV number | SNV的数目 |
| INDEL number | INDEL的数目 |
| SNV query | 在高置信基因组区域中的SNV数目 |
| INDEL query | 在高置信基因组区域中INDEL数目 |
| SNV TP | 真阳性SNV |
| INDEL TP | 真阳性INDEL |
| SNV FP | 假阳性SNV |
| INDEL FP | 假阳性INDEL |
| SNV FN | 假阴性SNV |
| INDEL FN | 假阴性INDEL |
| SNV precision | SNV与标准集比较的precision |
| INDEL precision | INDEL的与标准集比较的precision |
| SNV recall | SNV与标准集比较的recall |
| INDEL recall | INDEL的与标准集比较的recall |
| SNV F1 | SNV与标准集比较的F1-score |
| INDEL F1 | INDEL与标准集比较的F1-score |
与标准集比较的家庭单元整合结果reference_datasets_aver-std.txt
| Mean | SD | |
|---|---|---|
| SNV precision | ||
| INDEL precision | ||
| SNV recall | ||
| INDEL recall | ||
| SNV F1 | ||
| INDEL F1 |
2 Quartet家系关系评估 mendelian.txt
| 列名 | 说明 |
|---|---|
| Family | 家庭名字,我们目前的设计是4个Quartet样本,每个三个技术重复,family_1是指rep1的4个样本组成的家庭单位,以此类推。 |
| Total_Variants | 四个Quartet样本一共能检测到的变异位点数目 |
| Mendelian_Concordant_Variants | 符合孟德尔规律的变异位点数目 |
| Mendelian_Concordance_Quartet | 符合孟德尔遗传的比例 |
家系结果的整合结果quartet_indel_aver-std.txt和quartet_snv_aver-std.txt
| Mean | SD | |
|---|---|---|
| SNV/INDEL(根据文件名判断) |