
 LUYAO REN
3 years ago
LUYAO REN
3 years ago
commit
bd97ece857
60 changed files with 4289 additions and 0 deletions
+ 297
- 0
README.md
View File
| @@ -0,0 +1,297 @@ | |||
| # WES-germline Small Variants Quality Control Pipeline(Start from FASTQ files) | |||
| > Author: Run Luyao | |||
| > | |||
| > E-mail:18110700050@fudan.edu.cn | |||
| > | |||
| > Git: http://choppy.3steps.cn/renluyao/quartet-wes-germline-data-generation-qc.git | |||
| > | |||
| > Last Updates: 2021/04/14 | |||
| ## 安装指南 | |||
| ``` | |||
| # 激活choppy环境 | |||
| open-choppy-env | |||
| # 安装app | |||
| choppy install renluyao/quartet-wes-germline-data-generation-qc | |||
| ``` | |||
| ## App概述——中华家系1号标准物质介绍 | |||
| 建立高通量全基因组测序的生物计量和质量控制关键技术体系,是保障测序数据跨技术平台、跨实验室可比较、相关研究结果可重复、数据可共享的重要关键共性技术。建立国家基因组标准物质和基准数据集,突破基因组学的生物计量技术,是将测序技术转化成临床应用的重要环节与必经之路,目前国际上尚属空白。中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制了人源中华家系1号基因组标准物质(**Quartet,一套4个样本,编号分别为LCL5,LCL6,LCL7,LCL8,其中LCL5和LCL6为同卵双胞胎女儿,LCL7为父亲,LCL8为母亲**),以及相应的全基因组测序序列基准数据集(“量值”),为衡量基因序列检测准确与否提供一把“标尺”,成为保障基因测序数据可靠性的国家基准。人源中华家系1号基因组标准物质来源于泰州队列同卵双生双胞胎家庭,从遗传结构上体现了我国南北交界的人群结构特征,同时家系的设计也为“量值”的确定提供了遗传学依据。 | |||
| 中华家系1号DNA标准物质的Small Variants标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。此外,我们还可根据中华家系1号的生物遗传关系计算同卵双胞胎检测突变的一致性和符合四口之家遗传规律的一致率估计测序错误的比例,评估数据产生和分析的质量好坏。 | |||
| 2021年5-7月在临检中心的领导下组织全国临床和科研实验室全外显子测序室间质量评价预研https://www.nccl.org.cn/showEqaPtDetail?id=1514 | |||
|  | |||
| 该Quality_control APP用于全基因组测序(whole-exome sequencing,WES)数据的质量评估,包括原始数据质控、比对数据质控和突变检出数据质控。 | |||
| ## 流程与参数 | |||
|  | |||
|  | |||
| ### 1. 原始数据质量控制 | |||
| #### [Fastqc](<https://www.bioinformatics.babraham.ac.uk/projects/fastqc/>) v0.11.5 | |||
| FastQC是一个常用的测序原始数据的质控软件,主要包括12个模块,具体请参考[Fastqc模块详情](<https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/>)。 | |||
| ```bash | |||
| fastqc -t <threads> -o <output_directory> <fastq_file> | |||
| ``` | |||
| #### [Fastq Screen](<https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/>) 0.12.0 | |||
| Fastq Screen是检测测序原始数据中是否引⼊入其他物种,或是接头引物等污染,⽐比如,如果测序样本 | |||
| 是⼈人类,我们期望99%以上的reads匹配到⼈人类基因组,10%左右的reads匹配到与⼈人类基因组同源性 | |||
| 较⾼高的⼩小⿏鼠上。如果有过多的reads匹配到Ecoli或者Yeast,要考虑是否在培养细胞的时候细胞系被污 | |||
| 染,或者建库时⽂文库被污染。 | |||
| ```bash | |||
| fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file> | |||
| ``` | |||
| `--conf` conifg 文件主要输入了多个物种的fasta文件地址,可根据自己自己的需求下载其他物种的fasta文件加入分析 | |||
| `--top`一般不需要对整个fastq文件进行检索,取前100000行 | |||
| ### 2. 比对后数据质量控制 | |||
| #### [Qualimap](<http://qualimap.bioinfo.cipf.es/>) 2.0.0 | |||
| Qualimap是一个比对指控软件,包含Picard的MarkDuplicates的结果和sentieon中metrics的质控结果。 | |||
| ```bash | |||
| qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G | |||
| ``` | |||
| ### 3. 突变检出数据质量控制 | |||
| 突变质量控制的流程如下 | |||
|  | |||
| #### 3.1 根据标准数据集的数据质量控制 | |||
| #### [Hap.py](<https://github.com/Illumina/hap.py>) v0.3.9 | |||
| hap.py是将被检测vcf结果与benchmarking对比,计算precision和recall的软件,它考虑了vcf中[突变表示形式的多样性](<https://genome.sph.umich.edu/wiki/Variant_Normalization>),进行了归一化。 | |||
| ```bash | |||
| hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename> | |||
| ``` | |||
| #### 3.2 根据Quartet四口之家遗传规律的质量控制 | |||
| #### Reproducibility (in-house python script) | |||
| 标准数据集是根据我们整合多个平台方法,过滤不可重复检测、不符合孟德尔遗传规律的假阳性的突变。它可以评估数据产生和分析方法的相对好坏,但是具有一定的局限性,因为它排除掉了很多难测的基因组区域。我们可以通过比较同卵双胞胎突变检测的一致性对全基因组范围进行评估。 | |||
| #### [Mendelian Concordance Ratio](https://github.com/sbg/VBT-TrioAnalysis) (vbt v1.1) | |||
| 我们首先将四口之家拆分成两个三口之家进行孟德尔遗传的分析。当一个突变符合姐妹一致,且与父母符合孟德尔遗传规律,则认为是符合Quartet四口之家的孟德尔遗传规律。孟德尔符合率是指四个标准检测出的所有突变中满足孟德尔遗传规律的比例。 | |||
| ```bash | |||
| vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads> | |||
| ``` | |||
| ## App输入文件 | |||
| ```bash | |||
| choppy samples renluyao/quartet-wes-germline-data-generation-qc-latest --output samples | |||
| ``` | |||
| ####Samples文件的输入包括 | |||
| **1. inputSamplesFile**,该文件的上传至阿里云,samples文件中填写该文件的阿里云地址 | |||
| 请查看示例 **inputSamples.Examples.txt** | |||
| ```bash | |||
| #read1 #read2 #sample_name | |||
| ``` | |||
| read1 是阿里云上fastq read1的地址 | |||
| read2 是阿里云上fastq read2的地址 | |||
| sample_name是指样本的命名 | |||
| 所有上传的文件应有规范的命名 | |||
| Quartet_DNA_SequenceTech_SequenceMachine_SequenceSite_Sample_Replicate_Date.R1/R2.fastq.gz | |||
| SequenceTech是指测序平台,如ILM、BGI等 | |||
| SequenceMachine是指测序仪器,如XTen、Nova、Hiseq(Illumina)、SEQ500、SEQ1000(BGI)等 | |||
| SequenceSite是指测序单位的英文缩写 | |||
| Sample是指LCL5、LCL6、LCL7、LCL8 | |||
| Replicate是指技术重复,从1开始依次增加 | |||
| Date是指数据获得日期,格式为20200710 | |||
| 后缀一定是R1/R2.fastq.gz,不可以随意更改,R1/R2不可以写成r1/r2,fastq.gz不可以写成fq.gz | |||
| 各个缩写规范请见 https://fudan-pgx.yuque.com/docs/share/5baa851b-da97-47b9-b6c4-78f2b60595ab?# 《数据命名规范》 | |||
| **2. project** | |||
| 这个项目的名称,可以写自己可以识别的字符串,只能写英文和数字,不可以写中文 | |||
| **3. bed** | |||
| WES的捕获区域,建库试剂盒对应的BED文件 | |||
| **samples文件的示例请查看choppy_samples_example.csv** | |||
| #### Quartet样本的组合问题 | |||
| ##### 1. 没有测LCL5和LCL6,或者没有同时测LCL5和LCL6 | |||
| 只给出原始数据质控、比对数据质控、与标准集的比较 | |||
| ##### 2. 包含LCL5和LCL6同卵双胞胎的数据,但是父母的数据不全 | |||
| 只给出原始数据质控、比对数据质控、与标准集的比较、同卵双胞胎一致性 | |||
| ##### 3. 四个quartet样本都测了 | |||
| 给出所有结果原始数据质控、比对数据质控、与标准集的比较、同卵双胞胎一致性,符合孟德尔遗传比例 | |||
| **注意**:本app假设每个批次测的技术重复都一样,如batch 1测了LCL5、LCL6、LCL7和LCL8,batch 2 和batch 3也都测了这四个样本。本app不解决特别复杂的问题,例如batch1测了LCL5,LCL6,batch2测了LCL7和LCL8,本app只能给出原始数据质控、比对数据质控、与标准集的比较,不会把多个批次的数据合并计算孟德尔符合率和姐妹一致性。 | |||
| ## App输出文件 | |||
| 本计算会产生大量的中间结果,这里说明最后整合好的结果文件。两个tasks输出最终的结果: | |||
| #### 1. extract_tables.wdl | |||
| 原始结果质控 pre_alignment.txt | |||
| 比对结果指控 post_alignment.txt | |||
| 突变检出指控 variants.calling.qc.txt | |||
| 如果用户输入4个一组完整的家系样本则可以得到每个家庭单位的precision和recall的平均值,用于报告第一页的展示: | |||
| reference_datasets_aver-std.txt | |||
| #### 2. quartet_mendelian.wdl | |||
| 基于Quartet家系的质控 mendelian.txt | |||
| Quartet家系结果的平均值和SD值,用于报告第一页的展示 | |||
| quartet_indel_aver-std.txt | |||
| quartet_snv_aver-std.txt | |||
| #### 3. D5_D6.WDL | |||
| 如果用户没有完整输入一组家庭,但有同时有D5和D6的信息,我们可以计算同卵双胞胎检测出的突变一致性,但是这部分输出暂不整合至报告中。 | |||
| ${project}.sister.txt | |||
| ## 结果展示与解读 | |||
| ####1. 原始数据质量控制 | |||
| 原始数据质量控制主要通过考察测序数据的基本特征判断数据质量的好坏,比如数据量是否达到要求、reads的重复率是否过多、碱基质量、ATGC四种碱基的分布、GC含量、接头序列含量以及是否有其他物种的污染等等。 | |||
| FastQC和FastqScreen是两个常用的原始数据质量控制软件 | |||
| 总结表格 **pre_alignment.txt** | |||
| | 列名 | 说明 | | |||
| | ------------------------- | ------------------------------------ | | |||
| | Sample | 样本名,R1结尾为read1,R2结尾为read2 | | |||
| | %Dup | % Duplicate reads | | |||
| | %GC | Average % GC content | | |||
| | Total Sequences (million) | Total sequences | | |||
| | %Human | 比对到人类基因组的比例 | | |||
| | %EColi | 比对到大肠杆菌基因组的比例 | | |||
| | %Adapter | 比对到接头序列的比例 | | |||
| | %Vector | 比对到载体基因组的比例 | | |||
| | %rRNA | 比对到rRNA序列的比例 | | |||
| | %Virus | 比对到病毒基因组的比例 | | |||
| | %Yeast | 比对到酵母基因组的比例 | | |||
| | %Mitoch | 比对到线粒体序列的比例 | | |||
| | %No hits | 没有比对到以上基因组的比例 | | |||
| #### 2. 比对后数据质量控制 | |||
| 总结表格**post_alignment.txt** | |||
| | 列名 | 说明 | | |||
| | --------------------- | --------------------------------------------- | | |||
| | Sample | 样本名 | | |||
| | %Mapping | % mapped reads | | |||
| | %Mismatch Rate | Mapping error rate | | |||
| | Mendelian Insert Size | Median insert size(bp) | | |||
| | %Q20 | % bases >Q20 | | |||
| | %Q30 | % bases >Q30 | | |||
| | Mean Coverage | Mean deduped coverage | | |||
| | Median Coverage | Median deduped coverage | | |||
| | PCT_1X | Fraction of genome with at least 1x coverage | | |||
| | PCT_5X | Fraction of genome with at least 5x coverage | | |||
| | PCT_10X | Fraction of genome with at least 10x coverage | | |||
| | PCT_30X | Fraction of genome with at least 30x coverage | | |||
| ####3. 突变检出数据质量控制 | |||
| 具体信息**variants.calling.qc.txt** | |||
| | 列名 | 说明 | | |||
| | --------------- | ------------------------------ | | |||
| | Sample | 样本名 | | |||
| | SNV number | 检测到SNV的数目 | | |||
| | INDEL number | 检测到INDEL的数目 | | |||
| | SNV query | 在高置信基因组区域中的SNV数目 | | |||
| | INDEL query | 在高置信基因组区域中INDEL数目 | | |||
| | SNV TP | 真阳性SNV | | |||
| | INDEL TP | 真阳性INDEL | | |||
| | SNV FP | 假阳性SNV | | |||
| | INDEL FP | 假阳性INDEL | | |||
| | SNV FN | 假阴性SNV | | |||
| | INDEL FN | 假阴性INDEL | | |||
| | SNV precision | SNV与标准集比较的precision | | |||
| | INDEL precision | INDEL的与标准集比较的precision | | |||
| | SNV recall | SNV与标准集比较的recall | | |||
| | INDEL recall | INDEL的与标准集比较的recall | | |||
| | SNV F1 | SNV与标准集比较的F1-score | | |||
| | INDEL F1 | INDEL与标准集比较的F1-score | | |||
| 与标准集比较的家庭单元整合结果**reference_datasets_aver-std.txt** | |||
| | | Mean | SD | | |||
| | --------------- | ---- | ---- | | |||
| | SNV precision | | | | |||
| | INDEL precision | | | | |||
| | SNV recall | | | | |||
| | INDEL recall | | | | |||
| | SNV F1 | | | | |||
| | INDEL F1 | | | | |||
| ####4 Quartet家系关系评估mendelian.txt | |||
| | 列名 | 说明 | | |||
| | ----------------------------- | ------------------------------------------------------------ | | |||
| | Family | 家庭名字,我们目前的设计是4个Quartet样本,每个三个技术重复,family_1是指rep1的4个样本组成的家庭单位,以此类推。 | | |||
| | Total_Variants | 四个Quartet样本一共能检测到的变异位点数目 | | |||
| | Mendelian_Concordant_Variants | 符合孟德尔规律的变异位点数目 | | |||
| | Mendelian_Concordance_Quartet | 符合孟德尔遗传的比例 | | |||
| 家系结果的整合结果**quartet_indel_aver-std.txt**和**quartet_snv_aver-std.txt** | |||
| | | Mean | SD | | |||
| | --------------------------- | ---- | ---- | | |||
| | SNV/INDEL(根据文件名判断) | | | | |||
+ 2
- 0
choppy_samples_example.csv
View File
| @@ -0,0 +1,2 @@ | |||
| sample_id,inputSamplesFile,project,bed | |||
| 1,oss://pgx-result/renluyao/dataportal.test.small.txt,Quartet_DNA_ILM_XTen_NVG_20170531,oss://pgx-reference-data/bed/cbcga/S07604514_Padded.bed | |||
BIN
codescripts/.DS_Store
View File
+ 90
- 0
codescripts/D5_D6.py
View File
| @@ -0,0 +1,90 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to calculate reproducibility between Quartet_D5 and Quartet_D6s") | |||
| parser.add_argument('-sister', '--sister', type=str, help='sister.txt', required=True) | |||
| parser.add_argument('-project', '--project', type=str, help='project name', required=True) | |||
| args = parser.parse_args() | |||
| sister_file = args.sister | |||
| project_name = args.project | |||
| # output file | |||
| output_name = project_name + '.sister.reproducibility.txt' | |||
| output_file = open(output_name,'w') | |||
| # input files | |||
| sister_dat = pd.read_table(sister_file) | |||
| indel_sister_same = 0 | |||
| indel_sister_diff = 0 | |||
| snv_sister_same = 0 | |||
| snv_sister_diff = 0 | |||
| for row in sister_dat.itertuples(): | |||
| # snv indel | |||
| if ',' in row[4]: | |||
| alt = row[4].split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row[4]) | |||
| alt = alt_max | |||
| ref = row[3] | |||
| if len(ref) == 1 and alt == 1: | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| cate = 'INDEL' | |||
| # sister | |||
| if row[5] == row[6]: | |||
| if row[5] == './.': | |||
| mendelian = 'noInfo' | |||
| sister_count = "no" | |||
| elif row[5] == '0/0': | |||
| mendelian = 'Ref' | |||
| sister_count = "no" | |||
| else: | |||
| mendelian = '1' | |||
| sister_count = "yes_same" | |||
| else: | |||
| mendelian = '0' | |||
| if (row[5] == './.' or row[5] == '0/0') and (row[6] == './.' or row[6] == '0/0'): | |||
| sister_count = "no" | |||
| else: | |||
| sister_count = "yes_diff" | |||
| if cate == 'SNV': | |||
| if sister_count == 'yes_same': | |||
| snv_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| snv_sister_diff += 1 | |||
| else: | |||
| pass | |||
| elif cate == 'INDEL': | |||
| if sister_count == 'yes_same': | |||
| indel_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| indel_sister_diff += 1 | |||
| else: | |||
| pass | |||
| indel_sister = indel_sister_same/(indel_sister_same + indel_sister_diff) | |||
| snv_sister = snv_sister_same/(snv_sister_same + snv_sister_diff) | |||
| outcolumn = 'Project\tReproducibility_D5_D6\n' | |||
| indel_outResult = project_name + '.INDEL' + '\t' + str(indel_sister) + '\n' | |||
| snv_outResult = project_name + '.SNV' + '\t' + str(snv_sister) + '\n' | |||
| output_file.write(outcolumn) | |||
| output_file.write(indel_outResult) | |||
| output_file.write(snv_outResult) | |||
+ 37
- 0
codescripts/Indel_bed.py
View File
| @@ -0,0 +1,37 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/MIE/vcf/mutation_type',header=None) | |||
| outIndel = open(sys.argv[1],'w') | |||
| for row in mut.itertuples(): | |||
| if ',' in row._4: | |||
| alt_seq = row._4.split(',') | |||
| alt_len = [len(i) for i in alt_seq] | |||
| alt = max(alt_len) | |||
| else: | |||
| alt = len(row._4) | |||
| ref = row._3 | |||
| pos = row._2 | |||
| if len(ref) == 1 and alt == 1: | |||
| pass | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
+ 72
- 0
codescripts/bed_region.py
View File
| @@ -0,0 +1,72 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = mut = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/vcf/mutation_type',header=None) | |||
| vote = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/all_info/benchmark.vote.mendelian.txt',header=None) | |||
| merged_df = pd.merge(vote, mut, how='inner', left_on=[0,1], right_on = [0,1]) | |||
| outFile = open(sys.argv[1],'w') | |||
| outIndel = open(sys.argv[2],'w') | |||
| for row in merged_df.itertuples(): | |||
| #d5 | |||
| if ',' in row._7: | |||
| d5 = row._7.split(',') | |||
| d5_len = [len(i) for i in d5] | |||
| d5_alt = max(d5_len) | |||
| else: | |||
| d5_alt = len(row._7) | |||
| #d6 | |||
| if ',' in row._15: | |||
| d6 = row._15.split(',') | |||
| d6_len = [len(i) for i in d6] | |||
| d6_alt = max(d6_len) | |||
| else: | |||
| d6_alt = len(row._15) | |||
| #f7 | |||
| if ',' in row._23: | |||
| f7 = row._23.split(',') | |||
| f7_len = [len(i) for i in f7] | |||
| f7_alt = max(f7_len) | |||
| else: | |||
| f7_alt = len(row._23) | |||
| #m8 | |||
| if ',' in row._31: | |||
| m8 = row._31.split(',') | |||
| m8_len = [len(i) for i in m8] | |||
| m8_alt = max(m8_len) | |||
| else: | |||
| m8_alt = len(row._31) | |||
| all_length = [d5_alt,d6_alt,f7_alt,m8_alt] | |||
| alt = max(all_length) | |||
| ref = row._35 | |||
| pos = int(row._2) | |||
| if len(ref) == 1 and alt == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| outline = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\t' + str(row._2) + '\t' + cate + '\n' | |||
| outFile.write(outline) | |||
+ 67
- 0
codescripts/cluster.sh
View File
| @@ -0,0 +1,67 @@ | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$7"\t.\t.\t.\tGT\t"$6}' | grep -v '0/0' > LCL5.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$15"\t.\t.\t.\tGT\t"$14}' | grep -v '0/0' > LCL6.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$23"\t.\t.\t.\tGT\t"$22}' | grep -v '0/0'> LCL7.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$31"\t.\t.\t.\tGT\t"$30}'| grep -v '0/0' > LCL8.body | |||
| cat header5 LCL5.body > LCL5.beforediffbed.vcf | |||
| cat header6 LCL6.body > LCL6.beforediffbed.vcf | |||
| cat header7 LCL7.body > LCL7.beforediffbed.vcf | |||
| cat header8 LCL8.body > LCL8.beforediffbed.vcf | |||
| rtg bgzip *beforediffbed.vcf | |||
| rtg index *beforediffbed.vcf.gz | |||
| rtg vcffilter -i LCL5.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL5.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL6.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL6.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL7.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL7.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL8.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL8.afterfilterdiffbed.vcf.gz | |||
| /mnt/pgx_src_data_pool_4/home/renluyao/softwares/annovar/table_annovar.pl LCL5.beforediffbed.vcf.gz /mnt/pgx_src_data_pool_4/home/renluyao/softwares/annovar/humandb \ | |||
| -buildver hg38 \ | |||
| -out LCL5 \ | |||
| -remove \ | |||
| -protocol 1000g2015aug_all,1000g2015aug_afr,1000g2015aug_amr,1000g2015aug_eas,1000g2015aug_eur,1000g2015aug_sas,clinvar_20190305,gnomad211_genome \ | |||
| -operation f,f,f,f,f,f,f,f \ | |||
| -nastring . \ | |||
| -vcfinput \ | |||
| --thread 8 | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL5.afterfilterdiffbed.vcf.gz -o LCL5_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL6.afterfilterdiffbed.vcf.gz -o LCL6_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL7.afterfilterdiffbed.vcf.gz -o LCL7_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL8.afterfilterdiffbed.vcf.gz -o LCL8_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| bedtools subtract -a LCL5.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL5.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL6.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL6.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL7.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL7.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL8.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL8.27.homo_ref.consensus.filtereddiffbed.bed | |||
| python vcf2bed.py LCL5.body LCL5.variants.bed | |||
| python vcf2bed.py LCL6.body LCL6.variants.bed | |||
| python vcf2bed.py LCL7.body LCL7.variants.bed | |||
| python vcf2bed.py LCL8.body LCL8.variants.bed | |||
| cat /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/all_info/LCL5.variants.bed | cut -f1,11,12 | cat - LCL5.27.homo_ref.consensus.filtereddiffbed.bed | sort -k1,1 -k2,2n > LCL5.high.confidence.bed | |||
+ 156
- 0
codescripts/extract_tables.py
View File
| @@ -0,0 +1,156 @@ | |||
| import json | |||
| import pandas as pd | |||
| from functools import reduce | |||
| import sys, argparse, os | |||
| parser = argparse.ArgumentParser(description="This script is to get information from multiqc and sentieon, output the raw fastq, bam and variants calling (precision and recall) quality metrics") | |||
| parser.add_argument('-quality', '--quality_yield', type=str, help='*.quality_yield.txt', required=True) | |||
| parser.add_argument('-depth', '--wgs_metrics', type=str, help='*deduped_WgsMetricsAlgo.txt', required=True) | |||
| parser.add_argument('-aln', '--aln_metrics', type=str, help='*_deduped_aln_metrics.txt', required=True) | |||
| parser.add_argument('-is', '--is_metrics', type=str, help='*_deduped_is_metrics.txt', required=True) | |||
| parser.add_argument('-fastqc', '--fastqc', type=str, help='multiqc_fastqc.txt', required=True) | |||
| parser.add_argument('-fastqscreen', '--fastqscreen', type=str, help='multiqc_fastq_screen.txt', required=True) | |||
| parser.add_argument('-hap', '--happy', type=str, help='multiqc_happy_data.json', required=True) | |||
| parser.add_argument('-project', '--project_name', type=str, help='project_name', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| quality_yield_file = args.quality_yield | |||
| wgs_metrics_file = args.wgs_metrics | |||
| aln_metrics_file = args.aln_metrics | |||
| is_metrics_file = args.is_metrics | |||
| fastqc_file = args.fastqc | |||
| fastqscreen_file = args.fastqscreen | |||
| hap_file = args.happy | |||
| project_name = args.project_name | |||
| ############################################# | |||
| # fastqc | |||
| fastqc = pd.read_table(fastqc_file) | |||
| #fastqc = dat.loc[:, dat.columns.str.startswith('FastQC')] | |||
| #fastqc.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| #fastqc_stat = fastqc.dropna() | |||
| # qulimap | |||
| #qualimap = dat.loc[:, dat.columns.str.startswith('QualiMap')] | |||
| #qualimap.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| #qualimap_stat = qualimap.dropna() | |||
| # fastqc | |||
| #dat = pd.read_table(fastqc_file) | |||
| #fastqc_module = dat.loc[:, "per_base_sequence_quality":"kmer_content"] | |||
| #fastqc_module.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| #fastqc_all = pd.merge(fastqc_stat,fastqc_module, how='outer', left_on=['Sample'], right_on = ['Sample']) | |||
| # fastqscreen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| dat['Sample'] = [i.replace('_screen','') for i in dat['Sample']] | |||
| fastqscreen.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| # pre-alignment | |||
| pre_alignment_dat = pd.merge(fastqc,fastqscreen,how="outer",left_on=['Sample'],right_on=['Sample']) | |||
| pre_alignment_dat['FastQC_mqc-generalstats-fastqc-total_sequences'] = pre_alignment_dat['FastQC_mqc-generalstats-fastqc-total_sequences']/1000000 | |||
| del pre_alignment_dat['FastQC_mqc-generalstats-fastqc-percent_fails'] | |||
| del pre_alignment_dat['FastQC_mqc-generalstats-fastqc-avg_sequence_length'] | |||
| del pre_alignment_dat['ERCC percentage'] | |||
| del pre_alignment_dat['Phix percentage'] | |||
| del pre_alignment_dat['Mouse percentage'] | |||
| pre_alignment_dat = pre_alignment_dat.round(2) | |||
| pre_alignment_dat.columns = ['Sample','%Dup','%GC','Total Sequences (million)','%Human','%EColi','%Adapter','%Vector','%rRNA','%Virus','%Yeast','%Mitoch','%No hits'] | |||
| pre_alignment_dat.to_csv('pre_alignment.txt',sep="\t",index=0) | |||
| ############################ | |||
| dat = pd.read_table(aln_metrics_file,index_col=False) | |||
| dat['PCT_ALIGNED_READS'] = dat["PF_READS_ALIGNED"]/dat["TOTAL_READS"] | |||
| aln_metrics = dat[["Sample", "PCT_ALIGNED_READS","PF_MISMATCH_RATE"]] | |||
| aln_metrics = aln_metrics * 100 | |||
| aln_metrics['Sample'] = [x[-1] for x in aln_metrics['Sample'].str.split('/')] | |||
| dat = pd.read_table(is_metrics_file,index_col=False) | |||
| is_metrics = dat[['Sample', 'MEDIAN_INSERT_SIZE']] | |||
| is_metrics['Sample'] = [x[-1] for x in is_metrics['Sample'].str.split('/')] | |||
| dat = pd.read_table(quality_yield_file,index_col=False) | |||
| dat['%Q20'] = dat['Q20_BASES']/dat['TOTAL_BASES'] | |||
| dat['%Q30'] = dat['Q30_BASES']/dat['TOTAL_BASES'] | |||
| quality_yield = dat[['Sample','%Q20','%Q30']] | |||
| quality_yield = quality_yield * 100 | |||
| quality_yield['Sample'] = [x[-1] for x in quality_yield['Sample'].str.split('/')] | |||
| dat = pd.read_table(wgs_metrics_file,index_col=False) | |||
| wgs_metrics = dat[['Sample','MEAN_COVERAGE','MEDIAN_COVERAGE','PCT_1X', 'PCT_5X', 'PCT_10X','PCT_30X']] | |||
| wgs_metrics['PCT_1X'] = wgs_metrics['PCT_1X'] * 100 | |||
| wgs_metrics['PCT_5X'] = wgs_metrics['PCT_5X'] * 100 | |||
| wgs_metrics['PCT_10X'] = wgs_metrics['PCT_10X'] * 100 | |||
| wgs_metrics['PCT_30X'] = wgs_metrics['PCT_30X'] * 100 | |||
| wgs_metrics['Sample'] = [x[-1] for x in wgs_metrics['Sample'].str.split('/')] | |||
| data_frames = [aln_metrics, is_metrics, quality_yield, wgs_metrics] | |||
| post_alignment_dat = reduce(lambda left,right: pd.merge(left,right,on=['Sample'],how='outer'), data_frames) | |||
| post_alignment_dat.columns = ['Sample', '%Mapping', '%Mismatch Rate', 'Mendelian Insert Size','%Q20', '%Q30', 'Mean Coverage' ,'Median Coverage', 'PCT_1X', 'PCT_5X', 'PCT_10X','PCT_30X'] | |||
| post_alignment_dat = post_alignment_dat.round(2) | |||
| post_alignment_dat.to_csv('post_alignment.txt',sep="\t",index=0) | |||
| ######################################### | |||
| # variants calling | |||
| with open(hap_file) as hap_json: | |||
| happy = json.load(hap_json) | |||
| dat =pd.DataFrame.from_records(happy) | |||
| dat = dat.loc[:, dat.columns.str.endswith('ALL')] | |||
| dat_transposed = dat.T | |||
| dat_transposed = dat_transposed.loc[:,['sample_id','TRUTH.FN','QUERY.TOTAL','QUERY.FP','QUERY.UNK','METRIC.Precision','METRIC.Recall','METRIC.F1_Score']] | |||
| dat_transposed['QUERY.TP'] = dat_transposed['QUERY.TOTAL'].astype(int) - dat_transposed['QUERY.UNK'].astype(int) - dat_transposed['QUERY.FP'].astype(int) | |||
| dat_transposed['QUERY'] =dat_transposed['QUERY.TOTAL'].astype(int) - dat_transposed['QUERY.UNK'].astype(int) | |||
| indel = dat_transposed[['INDEL' in s for s in dat_transposed.index]] | |||
| snv = dat_transposed[['SNP' in s for s in dat_transposed.index]] | |||
| indel.reset_index(drop=True, inplace=True) | |||
| snv.reset_index(drop=True, inplace=True) | |||
| benchmark = pd.concat([snv, indel], axis=1) | |||
| benchmark = benchmark[["sample_id", 'QUERY.TOTAL', 'QUERY','QUERY.TP','QUERY.FP','TRUTH.FN','METRIC.Precision', 'METRIC.Recall','METRIC.F1_Score']] | |||
| benchmark.columns = ['Sample','sample_id','SNV number','INDEL number','SNV query','INDEL query','SNV TP','INDEL TP','SNV FP','INDEL FP','SNV FN','INDEL FN','SNV precision','INDEL precision','SNV recall','INDEL recall','SNV F1','INDEL F1'] | |||
| benchmark = benchmark[['Sample','SNV number','INDEL number','SNV query','INDEL query','SNV TP','INDEL TP','SNV FP','INDEL FP','SNV FN','INDEL FN','SNV precision','INDEL precision','SNV recall','INDEL recall','SNV F1','INDEL F1']] | |||
| benchmark['SNV precision'] = benchmark['SNV precision'].astype(float) | |||
| benchmark['INDEL precision'] = benchmark['INDEL precision'].astype(float) | |||
| benchmark['SNV recall'] = benchmark['SNV recall'].astype(float) | |||
| benchmark['INDEL recall'] = benchmark['INDEL recall'].astype(float) | |||
| benchmark['SNV F1'] = benchmark['SNV F1'].astype(float) | |||
| benchmark['INDEL F1'] = benchmark['INDEL F1'].astype(float) | |||
| benchmark = benchmark.round(2) | |||
| benchmark.to_csv('variants.calling.qc.txt',sep="\t",index=0) | |||
| #all_rep = [x.split('_')[6] for x in benchmark['Sample']] | |||
| #if all_rep.count(all_rep[0]) == 4: | |||
| # rep = list(set(all_rep)) | |||
| # columns = ['Family','Average Precision','Average Recall','Precison SD','Recall SD'] | |||
| # df_ = pd.DataFrame(columns=columns) | |||
| # for i in rep: | |||
| # string = "_" + i + "_" | |||
| # sub_dat = benchmark[benchmark['Sample'].str.contains('_1_')] | |||
| # mean = list(sub_dat.mean(axis = 0, skipna = True)) | |||
| # sd = list(sub_dat.std(axis = 0, skipna = True)) | |||
| # family_name = project_name + "." + i + ".SNV" | |||
| # df_ = df_.append({'Family': family_name, 'Average Precision': mean[0], 'Average Recall': mean[2], 'Precison SD': sd[0], 'Recall SD': sd[2] }, ignore_index=True) | |||
| # family_name = project_name + "." + i + ".INDEL" | |||
| # df_ = df_.append({'Family': family_name, 'Average Precision': mean[1], 'Average Recall': mean[3], 'Precison SD': sd[1], 'Recall SD': sd[3] }, ignore_index=True) | |||
| # df_ = df_.round(2) | |||
| # df_.to_csv('precision.recall.txt',sep="\t",index=0) | |||
| #else: | |||
| # pass | |||
+ 92
- 0
codescripts/extract_vcf_information.py
View File
| @@ -0,0 +1,92 @@ | |||
| import sys,getopt | |||
| import os | |||
| import re | |||
| import fileinput | |||
| import pandas as pd | |||
| def usage(): | |||
| print( | |||
| """ | |||
| Usage: python extract_vcf_information.py -i input_merged_vcf_file -o parsed_file | |||
| This script will extract SNVs and Indels information from the vcf files and output a tab-delimited files. | |||
| Input: | |||
| -i the selected vcf file | |||
| Output: | |||
| -o tab-delimited parsed file | |||
| """) | |||
| # select supported small variants | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| infoParsed = parse_INFO(strings[7]) | |||
| formatKeys = strings[8].split(':') | |||
| formatValues = strings[9].split(':') | |||
| for i in range(0,len(formatKeys) -1) : | |||
| if formatKeys[i] == 'AD': | |||
| ra = formatValues[i].split(',') | |||
| infoParsed['RefDP'] = ra[0] | |||
| infoParsed['AltDP'] = ra[1] | |||
| if (int(ra[1]) + int(ra[0])) != 0: | |||
| infoParsed['af'] = float(int(ra[1])/(int(ra[1]) + int(ra[0]))) | |||
| else: | |||
| pass | |||
| else: | |||
| infoParsed[formatKeys[i]] = formatValues[i] | |||
| infoParsed['chromo'] = strings[0] | |||
| infoParsed['pos'] = strings[1] | |||
| infoParsed['id'] = strings[2] | |||
| infoParsed['ref'] = strings[3] | |||
| infoParsed['alt'] = strings[4] | |||
| infoParsed['qual'] = strings[5] | |||
| return infoParsed | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| elif kv[0] == 'AF': | |||
| pass | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||
| for op,value in opts: | |||
| if op == "-i": | |||
| inputFile=value | |||
| elif op == "-o": | |||
| outputFile=value | |||
| elif op == "-h": | |||
| usage() | |||
| sys.exit() | |||
| if len(sys.argv[1:]) < 3: | |||
| usage() | |||
| sys.exit() | |||
| allDict = [] | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| oneDict = process(line) | |||
| allDict.append(oneDict) | |||
| allTable = pd.DataFrame(allDict) | |||
| allTable.to_csv(outputFile,sep='\t',index=False) | |||
+ 47
- 0
codescripts/filter_indel_over_50_cluster.py
View File
| @@ -0,0 +1,47 @@ | |||
| import sys,getopt | |||
| from itertools import islice | |||
| over_50_outfile = open("indel_lenth_over_50.txt",'w') | |||
| less_50_outfile = open("benchmark.men.vote.diffbed.lengthlessthan50.txt","w") | |||
| def process(line): | |||
| strings = line.strip().split('\t') | |||
| #d5 | |||
| if ',' in strings[6]: | |||
| d5 = strings[6].split(',') | |||
| d5_len = [len(i) for i in d5] | |||
| d5_alt = max(d5_len) | |||
| else: | |||
| d5_alt = len(strings[6]) | |||
| #d6 | |||
| if ',' in strings[14]: | |||
| d6 = strings[14].split(',') | |||
| d6_len = [len(i) for i in d6] | |||
| d6_alt = max(d6_len) | |||
| else: | |||
| d6_alt = len(strings[14]) | |||
| #f7 | |||
| if ',' in strings[22]: | |||
| f7 = strings[22].split(',') | |||
| f7_len = [len(i) for i in f7] | |||
| f7_alt = max(f7_len) | |||
| else: | |||
| f7_alt = len(strings[22]) | |||
| #m8 | |||
| if ',' in strings[30]: | |||
| m8 = strings[30].split(',') | |||
| m8_len = [len(i) for i in m8] | |||
| m8_alt = max(m8_len) | |||
| else: | |||
| m8_alt = len(strings[30]) | |||
| #ref | |||
| ref_len = len(strings[34]) | |||
| if (d5_alt > 50) or (d6_alt > 50) or (f7_alt > 50) or (m8_alt > 50) or (ref_len > 50): | |||
| over_50_outfile.write(line) | |||
| else: | |||
| less_50_outfile.write(line) | |||
| input_file = open(sys.argv[1]) | |||
| for line in islice(input_file, 1, None): | |||
| process(line) | |||
+ 43
- 0
codescripts/filter_indel_over_50_mendelian.py
View File
| @@ -0,0 +1,43 @@ | |||
| from itertools import islice | |||
| import fileinput | |||
| import sys, argparse, os | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to exclude indel over 50bp") | |||
| parser.add_argument('-i', '--mergedGVCF', type=str, help='merged gVCF txt with only chr, pos, ref, alt and genotypes', required=True) | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='prefix of output file', required=True) | |||
| args = parser.parse_args() | |||
| input_dat = args.mergedGVCF | |||
| prefix = args.prefix | |||
| # output file | |||
| output_name = prefix + '.indel.lessthan50bp.txt' | |||
| outfile = open(output_name,'w') | |||
| def process(line): | |||
| strings = line.strip().split('\t') | |||
| #d5 | |||
| if ',' in strings[3]: | |||
| alt = strings[3].split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(strings[3]) | |||
| #ref | |||
| ref_len = len(strings[2]) | |||
| if (alt_max > 50) or (ref_len > 50): | |||
| pass | |||
| else: | |||
| outfile.write(line) | |||
| for line in fileinput.input(input_dat): | |||
| process(line) | |||
+ 416
- 0
codescripts/high_confidence_call_vote.py
View File
| @@ -0,0 +1,416 @@ | |||
| from __future__ import division | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to count voting number") | |||
| parser.add_argument('-vcf', '--multi_sample_vcf', type=str, help='The VCF file you want to count the voting number', required=True) | |||
| parser.add_argument('-dup', '--dup_list', type=str, help='Duplication list', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='Prefix of output file name', required=True) | |||
| args = parser.parse_args() | |||
| multi_sample_vcf = args.multi_sample_vcf | |||
| dup_list = args.dup_list | |||
| prefix = args.prefix | |||
| sample_name = args.sample_name | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Sum depth of all samples"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Sum alternative depth of all samples"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency, sum alternative depth / sum depth"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Average genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Average mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUALI,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| vcf_header_all_sample = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DUP,Number=1,Type=Flag,Description="Duplicated variant records"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##INFO=<ID=ALL_ALT,Number=1,Type=Float,Description="Sum of alternative reads of all samples"> | |||
| ##INFO=<ID=ALL_DP,Number=1,Type=Float,Description="Sum of depth of all samples"> | |||
| ##INFO=<ID=ALL_AF,Number=1,Type=Float,Description="Allele frequency of net alternatice reads and net depth"> | |||
| ##INFO=<ID=GQ_MEAN,Number=1,Type=Float,Description="Mean of genotype quality of all samples"> | |||
| ##INFO=<ID=QD_MEAN,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##INFO=<ID=MQ_MEAN,Number=1,Type=Float,Description="Mean of mapping quality of all samples"> | |||
| ##INFO=<ID=FS_MEAN,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##INFO=<ID=QUAL_MEAN,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##INFO=<ID=PCR,Number=1,Type=String,Description="Consensus of PCR votes"> | |||
| ##INFO=<ID=PCR_FREE,Number=1,Type=String,Description="Consensus of PCR-free votes"> | |||
| ##INFO=<ID=CONSENSUS,Number=1,Type=String,Description="Consensus calls"> | |||
| ##INFO=<ID=CONSENSUS_SEQ,Number=1,Type=String,Description="Consensus sequence"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=String,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=String,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=String,Description="Genotype quality"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=String,Description="Mapping quality"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=String,Description="1 is twins shared, 0 is twins discordant "> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL5 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL6 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # read in duplication list | |||
| dup = pd.read_table(dup_list,header=None) | |||
| var_dup = dup[0].tolist() | |||
| # output file | |||
| benchmark_file_name = prefix + '_voted.vcf' | |||
| benchmark_outfile = open(benchmark_file_name,'w') | |||
| all_sample_file_name = prefix + '_all_sample_information.vcf' | |||
| all_sample_outfile = open(all_sample_file_name,'w') | |||
| # write VCF | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '_benchmark_calls\n' | |||
| benchmark_outfile.write(vcf_header) | |||
| benchmark_outfile.write(outputcolumn) | |||
| outputcolumn_all_sample = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+ \ | |||
| 'Quartet_DNA_BGI_SEQ2000_BGI_1_20180518\tQuartet_DNA_BGI_SEQ2000_BGI_2_20180530\tQuartet_DNA_BGI_SEQ2000_BGI_3_20180530\t' + \ | |||
| 'Quartet_DNA_BGI_T7_WGE_1_20191105\tQuartet_DNA_BGI_T7_WGE_2_20191105\tQuartet_DNA_BGI_T7_WGE_3_20191105\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_1_20181108\tQuartet_DNA_ILM_Nova_ARD_2_20181108\tQuartet_DNA_ILM_Nova_ARD_3_20181108\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_4_20190111\tQuartet_DNA_ILM_Nova_ARD_5_20190111\tQuartet_DNA_ILM_Nova_ARD_6_20190111\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_BRG_1_20180930\tQuartet_DNA_ILM_Nova_BRG_2_20180930\tQuartet_DNA_ILM_Nova_BRG_3_20180930\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_WUX_1_20190917\tQuartet_DNA_ILM_Nova_WUX_2_20190917\tQuartet_DNA_ILM_Nova_WUX_3_20190917\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_ARD_1_20170403\tQuartet_DNA_ILM_XTen_ARD_2_20170403\tQuartet_DNA_ILM_XTen_ARD_3_20170403\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_NVG_1_20170329\tQuartet_DNA_ILM_XTen_NVG_2_20170329\tQuartet_DNA_ILM_XTen_NVG_3_20170329\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_WUX_1_20170216\tQuartet_DNA_ILM_XTen_WUX_2_20170216\tQuartet_DNA_ILM_XTen_WUX_3_20170216\n' | |||
| all_sample_outfile.write(vcf_header_all_sample) | |||
| all_sample_outfile.write(outputcolumn_all_sample) | |||
| #function | |||
| def replace_nan(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| updated_list.append('.:.:.:.:.:.:.:.:.:.:.:.') | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def remove_dot(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| pass | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def detected_number(strings): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| percentage = 27 - gt.count('.') | |||
| return(str(percentage)) | |||
| def vote_number(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| vote_num = gt.count(consensus_call) | |||
| return(str(vote_num)) | |||
| def family_vote(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| indices = [i for i, x in enumerate(gt) if x == consensus_call] | |||
| matched_mendelian = itemgetter(*indices)(mendelian) | |||
| mendelian_num = matched_mendelian.count('1:1:1') | |||
| return(str(mendelian_num)) | |||
| def gt_uniform(strings): | |||
| uniformed_gt = '' | |||
| allele1 = strings.split('/')[0] | |||
| allele2 = strings.split('/')[1] | |||
| if int(allele1) > int(allele2): | |||
| uniformed_gt = allele2 + '/' + allele1 | |||
| else: | |||
| uniformed_gt = allele1 + '/' + allele2 | |||
| return uniformed_gt | |||
| def decide_by_rep(strings): | |||
| consensus_rep = '' | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| # modified gt turn 2/1 to 1/2 | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| # mendelian consistent? | |||
| mendelian_dict = Counter(mendelian) | |||
| highest_mendelian = mendelian_dict.most_common(1) | |||
| candidate_mendelian = highest_mendelian[0][0] | |||
| freq_mendelian = highest_mendelian[0][1] | |||
| if (candidate_mendelian == '1:1:1') and (freq_mendelian >= 2): | |||
| gt_num_dict = Counter(gt) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if (candidate_gt != '0/0') and (freq_gt >= 2): | |||
| consensus_rep = candidate_gt | |||
| elif (candidate_gt == '0/0') and (freq_gt >= 2): | |||
| consensus_rep = '0/0' | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| elif (candidate_mendelian == '') and (freq_mendelian >= 2): | |||
| consensus_rep = 'noInfo' | |||
| else: | |||
| consensus_rep = 'inconMen' | |||
| return consensus_rep | |||
| def main(): | |||
| for line in fileinput.input(multi_sample_vcf): | |||
| headline = re.match('^\#',line) | |||
| if headline is not None: | |||
| pass | |||
| else: | |||
| line = line.strip() | |||
| strings = line.split('\t') | |||
| variant_id = '_'.join([strings[0],strings[1]]) | |||
| # check if the variants location is duplicated | |||
| if variant_id in var_dup: | |||
| strings[7] = strings[7] + ';DUP' | |||
| outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(outLine) | |||
| else: | |||
| # pre-define | |||
| pcr_consensus = '.' | |||
| pcr_free_consensus = '.' | |||
| consensus_call = '.' | |||
| consensus_alt_seq = '.' | |||
| # pcr | |||
| strings[9:] = replace_nan(strings[9:]) | |||
| pcr = itemgetter(*[9,10,11,27,28,29,30,31,32,33,34,35])(strings) | |||
| SEQ2000 = decide_by_rep(pcr[0:3]) | |||
| XTen_ARD = decide_by_rep(pcr[3:6]) | |||
| XTen_NVG = decide_by_rep(pcr[6:9]) | |||
| XTen_WUX = decide_by_rep(pcr[9:12]) | |||
| sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 2: | |||
| pcr_consensus = candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| pcr_free = itemgetter(*[12,13,14,15,16,17,18,19,20,21,22,23,24,25,26])(strings) | |||
| T7_WGE = decide_by_rep(pcr_free[0:3]) | |||
| Nova_ARD_1 = decide_by_rep(pcr_free[3:6]) | |||
| Nova_ARD_2 = decide_by_rep(pcr_free[6:9]) | |||
| Nova_BRG = decide_by_rep(pcr_free[9:12]) | |||
| Nova_WUX = decide_by_rep(pcr_free[12:15]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG,Nova_WUX] | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 3: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| # pcr and pcr-free | |||
| tag = ['inconGT','noInfo','inconMen','inconSequenceSite'] | |||
| if (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag) and (pcr_consensus != '0/0'): | |||
| consensus_call = pcr_consensus | |||
| VOTED = vote_number(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';VOTED=' + VOTED | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| FAM = family_vote(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';FAM=' + FAM | |||
| # Delete multiple alternative genotype to necessary expression | |||
| alt = strings[4] | |||
| alt_gt = alt.split(',') | |||
| if len(alt_gt) > 1: | |||
| allele1 = consensus_call.split('/')[0] | |||
| allele2 = consensus_call.split('/')[1] | |||
| if allele1 == '0': | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| consensus_alt_seq = allele2_seq | |||
| consensus_call = '0/1' | |||
| else: | |||
| allele1_seq = alt_gt[int(allele1) - 1] | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| if int(allele1) > int(allele2): | |||
| consensus_alt_seq = allele2_seq + ',' + allele1_seq | |||
| consensus_call = '1/2' | |||
| elif int(allele1) < int(allele2): | |||
| consensus_alt_seq = allele1_seq + ',' + allele2_seq | |||
| consensus_call = '1/2' | |||
| else: | |||
| consensus_alt_seq = allele1_seq | |||
| consensus_call = '1/1' | |||
| else: | |||
| consensus_alt_seq = alt | |||
| # GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL | |||
| # GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT | |||
| # DP | |||
| DP = [x.split(':')[4] for x in strings[9:]] | |||
| DP = remove_dot(DP) | |||
| DP = [int(x) for x in DP] | |||
| ALL_DP = sum(DP) | |||
| # AF | |||
| ALT = [x.split(':')[5] for x in strings[9:]] | |||
| ALT = remove_dot(ALT) | |||
| ALT = [int(x) for x in ALT] | |||
| ALL_ALT = sum(ALT) | |||
| ALL_AF = round(ALL_ALT/ALL_DP,2) | |||
| # GQ | |||
| GQ = [x.split(':')[7] for x in strings[9:]] | |||
| GQ = remove_dot(GQ) | |||
| GQ = [int(x) for x in GQ] | |||
| GQ_MEAN = round(mean(GQ),2) | |||
| # QD | |||
| QD = [x.split(':')[8] for x in strings[9:]] | |||
| QD = remove_dot(QD) | |||
| QD = [float(x) for x in QD] | |||
| QD_MEAN = round(mean(QD),2) | |||
| # MQ | |||
| MQ = [x.split(':')[9] for x in strings[9:]] | |||
| MQ = remove_dot(MQ) | |||
| MQ = [float(x) for x in MQ] | |||
| MQ_MEAN = round(mean(MQ),2) | |||
| # FS | |||
| FS = [x.split(':')[10] for x in strings[9:]] | |||
| FS = remove_dot(FS) | |||
| FS = [float(x) for x in FS] | |||
| FS_MEAN = round(mean(FS),2) | |||
| # QUAL | |||
| QUAL = [x.split(':')[11] for x in strings[9:]] | |||
| QUAL = remove_dot(QUAL) | |||
| QUAL = [float(x) for x in QUAL] | |||
| QUAL_MEAN = round(mean(QUAL),2) | |||
| # benchmark output | |||
| output_format = consensus_call + ':' + str(ALL_DP) + ':' + str(ALL_ALT) + ':' + str(ALL_AF) + ':' + str(GQ_MEAN) + ':' + str(QD_MEAN) + ':' + str(MQ_MEAN) + ':' + str(FS_MEAN) + ':' + str(QUAL_MEAN) | |||
| outLine = strings[0] + '\t' + strings[1] + '\t' + strings[2] + '\t' + strings[3] + '\t' + consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' + strings[7] + '\t' + 'GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + output_format + '\n' | |||
| benchmark_outfile.write(outLine) | |||
| # all sample output | |||
| strings[7] = strings[7] + ';ALL_ALT=' + str(ALL_ALT) + ';ALL_DP=' + str(ALL_DP) + ';ALL_AF=' + str(ALL_AF) \ | |||
| + ';GQ_MEAN=' + str(GQ_MEAN) + ';QD_MEAN=' + str(QD_MEAN) + ';MQ_MEAN=' + str(MQ_MEAN) + ';FS_MEAN=' + str(FS_MEAN) \ | |||
| + ';QUAL_MEAN=' + str(QUAL_MEAN) + ';PCR=' + consensus_call + ';PCR_FREE=' + consensus_call + ';CONSENSUS=' + consensus_call \ | |||
| + ';CONSENSUS_SEQ=' + consensus_alt_seq | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus == '0/0') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != '0/0')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus != '0/0') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == '0/0')): | |||
| consensus_call = 'pcr-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call + ';PCR=' + pcr_consensus + ';PCR_FREE=' + pcr_free_consensus | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = 'confirm for parents' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| else: | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| if __name__ == '__main__': | |||
| main() | |||
+ 42
- 0
codescripts/high_voted_mendelian_bed.py
View File
| @@ -0,0 +1,42 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = pd.read_table(sys.argv[1]) | |||
| outFile = open(sys.argv[2],'w') | |||
| for row in mut.itertuples(): | |||
| #d5 | |||
| if ',' in row.V4: | |||
| alt = row.V4.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.V4) | |||
| #d6 | |||
| alt = alt_max | |||
| ref = row.V3 | |||
| pos = int(row.V2) | |||
| if len(ref) == 1 and alt == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline = row.V1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\t' + str(row.V2) + '\t' + cate + '\n' | |||
| outFile.write(outline) | |||
+ 50
- 0
codescripts/how_many_samples.py
View File
| @@ -0,0 +1,50 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| from operator import itemgetter | |||
| parser = argparse.ArgumentParser(description="This script is to get how many samples") | |||
| parser.add_argument('-sample', '--sample', type=str, help='quartet_sample', required=True) | |||
| parser.add_argument('-rep', '--rep', type=str, help='quartet_rep', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| sample = args.sample | |||
| rep = args.rep | |||
| quartet_sample = pd.read_table(sample,header=None) | |||
| quartet_sample = list(quartet_sample[0]) | |||
| quartet_rep = pd.read_table(rep.header=None) | |||
| quartet_rep = quartet_rep[0] | |||
| #tags | |||
| sister_tag = 'false' | |||
| quartet_tag = 'false' | |||
| quartet_rep_unique = list(set(quartet_rep)) | |||
| single_rep = [i for i in range(len(quartet_rep)) if quartet_rep[i] == quartet_rep_unique[0]] | |||
| single_batch_sample = itemgetter(*single_rep)(quartet_sample) | |||
| num = len(single_batch_sample) | |||
| if num == 1: | |||
| sister_tag = 'false' | |||
| quartet_tag = 'false' | |||
| elif num == 2: | |||
| if set(single_batch_sample) == set(['LCL5','LCL6']): | |||
| sister_tag = 'true' | |||
| quartet_tag = 'false' | |||
| elif num == 3: | |||
| if ('LCL5' in single_batch_sample) and ('LCL6' in single_batch_sample): | |||
| sister_tag = 'true' | |||
| quartet_tag = 'false' | |||
| elif num == 4: | |||
| if set(single_batch_sample) == set(['LCL5','LCL6','LCL7','LCL8']): | |||
| sister_tag = 'false' | |||
| quartet_tag = 'true' | |||
| sister_outfile = open('sister_tag','w') | |||
| quartet_outfile = open('quartet_tag','w') | |||
| sister_outfile.write(sister_tag) | |||
| quartet_outfile.write(quartet_tag) | |||
+ 42
- 0
codescripts/lcl5_all_called_variants.py
View File
| @@ -0,0 +1,42 @@ | |||
| from __future__ import division | |||
| import sys, argparse, os | |||
| import pandas as pd | |||
| from collections import Counter | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| vcf = args.vcf | |||
| lcl5_outfile = open('LCL5_all_variants.txt','w') | |||
| filtered_outfile = open('LCL5_filtered_variants.txt','w') | |||
| vcf_dat = pd.read_table(vcf) | |||
| for row in vcf_dat.itertuples(): | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl5_vcf_gt = [x.split(':')[0] for x in lcl5_list] | |||
| lcl5_gt=[item.replace('./.', '0/0') for item in lcl5_vcf_gt] | |||
| gt_dict = Counter(lcl5_gt) | |||
| highest_gt = gt_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| output = row._1 + '\t' + str(row.POS) + '\t' + '\t'.join(lcl5_gt) + '\n' | |||
| if (candidate_gt == '0/0') and (freq_gt == 27): | |||
| filtered_outfile.write(output) | |||
| else: | |||
| lcl5_outfile.write(output) | |||
+ 6
- 0
codescripts/linux_command.sh
View File
| @@ -0,0 +1,6 @@ | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$7"\t.\t.\t.\tGT\t"$6}' | grep -v '2_y' > LCL5.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$15"\t.\t.\t.\tGT\t"$14}' | grep -v '2_y' > LCL6.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$23"\t.\t.\t.\tGT\t"$22}' | grep -v '2_y' > LCL7.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$31"\t.\t.\t.\tGT\t"$30}' | grep -v '2_y' > LCL8.body | |||
| for i in *txt; do cat $i | awk '{ if ((length($3) == 1) && (length($4) == 1)) { print } }' | grep -v '#' | cut -f3,4 | sort |uniq -c | sed 's/\s\+/\t/g' | cut -f2 > $i.mut; done | |||
+ 129
- 0
codescripts/merge_mendelian_vcfinfo.py
View File
| @@ -0,0 +1,129 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to get final high confidence calls and information of all replicates") | |||
| parser.add_argument('-vcfInfo', '--vcfInfo', type=str, help='The txt file of variants information, this file is named as prefix__variant_quality_location.txt', required=True) | |||
| parser.add_argument('-mendelianInfo', '--mendelianInfo', type=str, help='The merged mendelian information of all samples', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| args = parser.parse_args() | |||
| vcfInfo = args.vcfInfo | |||
| mendelianInfo = args.mendelianInfo | |||
| sample_name = args.sample_name | |||
| #GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=Flag,Description="1 for sister consistent, 0 for sister different"> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL5 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL6 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUAL,Number=1,Type=Float,Description="variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # output file | |||
| file_name = sample_name + '_mendelian_vcfInfo.vcf' | |||
| outfile = open(file_name,'w') | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '\n' | |||
| outfile.write(vcf_header) | |||
| outfile.write(outputcolumn) | |||
| # input files | |||
| vcf_info = pd.read_table(vcfInfo) | |||
| mendelian_info = pd.read_table(mendelianInfo) | |||
| merged_df = pd.merge(vcf_info, mendelian_info, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_df = merged_df.fillna('.') | |||
| # | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| # | |||
| for row in merged_df.itertuples(): | |||
| if row[18] != '.': | |||
| # format | |||
| # GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = row[18] + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_y + '\t' + row.ALT_y + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + FORMAT + '\n' | |||
| else: | |||
| rawGT = row[10].split(':') | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = '.:.:.:.' + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) + ':' + rawGT[0] | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_x + '\t' + row.ALT_x + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT' + '\t' + FORMAT + '\n' | |||
| outfile.write(outline) | |||
+ 71
- 0
codescripts/merge_two_family.py
View File
| @@ -0,0 +1,71 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to extract mendelian concordance information") | |||
| parser.add_argument('-LCL5', '--LCL5', type=str, help='LCL5 family info', required=True) | |||
| parser.add_argument('-LCL6', '--LCL6', type=str, help='LCL6 family info', required=True) | |||
| parser.add_argument('-family', '--family', type=str, help='family name', required=True) | |||
| args = parser.parse_args() | |||
| lcl5 = args.LCL5 | |||
| lcl6 = args.LCL6 | |||
| family = args.family | |||
| # output file | |||
| family_name = family + '.txt' | |||
| family_file = open(family_name,'w') | |||
| # input files | |||
| lcl5_dat = pd.read_table(lcl5) | |||
| lcl6_dat = pd.read_table(lcl6) | |||
| merged_df = pd.merge(lcl5_dat, lcl6_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| def alt_seq(alt, genotype): | |||
| if genotype == './.': | |||
| seq = './.' | |||
| elif genotype == '0/0': | |||
| seq = '0/0' | |||
| else: | |||
| alt = alt.split(',') | |||
| genotype = genotype.split('/') | |||
| if genotype[0] == '0': | |||
| allele2 = alt[int(genotype[1]) - 1] | |||
| seq = '0/' + allele2 | |||
| else: | |||
| allele1 = alt[int(genotype[0]) - 1] | |||
| allele2 = alt[int(genotype[1]) - 1] | |||
| seq = allele1 + '/' + allele2 | |||
| return seq | |||
| for row in merged_df.itertuples(): | |||
| # correction of multiallele | |||
| if pd.isnull(row.INFO_x) == True or pd.isnull(row.INFO_y) == True: | |||
| mendelian = '.' | |||
| else: | |||
| lcl5_seq = alt_seq(row.ALT_x, row.CHILD_x) | |||
| lcl6_seq = alt_seq(row.ALT_y, row.CHILD_y) | |||
| if lcl5_seq == lcl6_seq: | |||
| mendelian = '1' | |||
| else: | |||
| mendelian = '0' | |||
| if pd.isnull(row.INFO_x) == True: | |||
| mendelian = mendelian + ':.' | |||
| else: | |||
| mendelian = mendelian + ':' + row.INFO_x.split('=')[1] | |||
| if pd.isnull(row.INFO_y) == True: | |||
| mendelian = mendelian + ':.' | |||
| else: | |||
| mendelian = mendelian + ':' + row.INFO_y.split('=')[1] | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + mendelian + '\n' | |||
| family_file.write(outline) | |||
+ 161
- 0
codescripts/merge_two_family_with_genotype.py
View File
| @@ -0,0 +1,161 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to extract mendelian concordance information") | |||
| parser.add_argument('-LCL5', '--LCL5', type=str, help='LCL5 family info', required=True) | |||
| parser.add_argument('-LCL6', '--LCL6', type=str, help='LCL6 family info', required=True) | |||
| parser.add_argument('-genotype', '--genotype', type=str, help='Genotype information of a set of four family members', required=True) | |||
| parser.add_argument('-family', '--family', type=str, help='family name', required=True) | |||
| args = parser.parse_args() | |||
| lcl5 = args.LCL5 | |||
| lcl6 = args.LCL6 | |||
| genotype = args.genotype | |||
| family = args.family | |||
| # output file | |||
| family_name = family + '.txt' | |||
| family_file = open(family_name,'w') | |||
| summary_name = family + '.summary.txt' | |||
| summary_file = open(summary_name,'w') | |||
| # input files | |||
| lcl5_dat = pd.read_table(lcl5) | |||
| lcl6_dat = pd.read_table(lcl6) | |||
| genotype_dat = pd.read_table(genotype) | |||
| merged_df = pd.merge(lcl5_dat, lcl6_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_genotype_df = pd.merge(merged_df, genotype_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_genotype_df = merged_genotype_df.dropna() | |||
| merged_genotype_df_sub = merged_genotype_df.iloc[:,[0,1,23,24,29,30,31,32,7,17]] | |||
| merged_genotype_df_sub.columns = ['CHROM', 'POS', 'REF', 'ALT','LCL5','LCL6','LCL7','LCL8', 'TRIO5', 'TRIO6'] | |||
| indel_sister_same = 0 | |||
| indel_sister_diff = 0 | |||
| indel_family_all = 0 | |||
| indel_family_mendelian = 0 | |||
| snv_sister_same = 0 | |||
| snv_sister_diff = 0 | |||
| snv_family_all = 0 | |||
| snv_family_mendelian = 0 | |||
| for row in merged_genotype_df_sub.itertuples(): | |||
| # indel or snv | |||
| if ',' in row.ALT: | |||
| alt = row.ALT.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.ALT) | |||
| alt = alt_max | |||
| ref = row.REF | |||
| if len(ref) == 1 and alt == 1: | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| cate = 'INDEL' | |||
| # sister | |||
| if row.LCL5 == row.LCL6: | |||
| if row.LCL5 == './.': | |||
| mendelian = 'noInfo' | |||
| sister_count = "no" | |||
| elif row.LCL5 == '0/0': | |||
| mendelian = 'Ref' | |||
| sister_count = "no" | |||
| else: | |||
| mendelian = '1' | |||
| sister_count = "yes_same" | |||
| else: | |||
| mendelian = '0' | |||
| if (row.LCL5 == './.' or row.LCL5 == '0/0') and (row.LCL6 == './.' or row.LCL6 == '0/0'): | |||
| sister_count = "no" | |||
| else: | |||
| sister_count = "yes_diff" | |||
| # family trio5 | |||
| if row.LCL5 == row.LCL7 == row.LCL8 == './.': | |||
| mendelian = mendelian + ':noInfo' | |||
| elif row.LCL5 == row.LCL7 == row.LCL8 == '0/0': | |||
| mendelian = mendelian + ':Ref' | |||
| elif pd.isnull(row.TRIO5) == True: | |||
| mendelian = mendelian + ':unVBT' | |||
| else: | |||
| mendelian = mendelian + ':' + row.TRIO5.split('=')[1] | |||
| # family trio6 | |||
| if row.LCL6 == row.LCL7 == row.LCL8 == './.': | |||
| mendelian = mendelian + ':noInfo' | |||
| elif row.LCL6 == row.LCL7 == row.LCL8 == '0/0': | |||
| mendelian = mendelian + ':Ref' | |||
| elif pd.isnull(row.TRIO6) == True: | |||
| mendelian = mendelian + ':unVBT' | |||
| else: | |||
| mendelian = mendelian + ':' + row.TRIO6.split('=')[1] | |||
| # not count into family | |||
| if (row.LCL5 == './.' or row.LCL5 == '0/0') and (row.LCL6 == './.' or row.LCL6 == '0/0') and (row.LCL7 == './.' or row.LCL7 == '0/0') and (row.LCL8 == './.' or row.LCL8 == '0/0'): | |||
| mendelian_count = "no" | |||
| else: | |||
| mendelian_count = "yes" | |||
| outline = str(row.CHROM) + '\t' + str(row.POS) + '\t' + str(row.REF) + '\t' + str(row.ALT) + '\t' + str(cate) + '\t' + str(row.LCL5) + '\t' + str(row.LCL6) + '\t' + str(row.LCL7) + '\t' + str(row.LCL8) + '\t' + str(row.TRIO5) + '\t' + str(row.TRIO6) + '\t' + str(mendelian) + '\t' + str(mendelian_count) + '\t' + str(sister_count) + '\n' | |||
| family_file.write(outline) | |||
| if cate == 'SNV': | |||
| if sister_count == 'yes_same': | |||
| snv_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| snv_sister_diff += 1 | |||
| else: | |||
| pass | |||
| if mendelian_count == 'yes': | |||
| snv_family_all += 1 | |||
| else: | |||
| pass | |||
| if mendelian == '1:1:1': | |||
| snv_family_mendelian += 1 | |||
| elif mendelian == 'Ref:1:1': | |||
| snv_family_mendelian += 1 | |||
| else: | |||
| pass | |||
| elif cate == 'INDEL': | |||
| if sister_count == 'yes_same': | |||
| indel_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| indel_sister_diff += 1 | |||
| else: | |||
| pass | |||
| if mendelian_count == 'yes': | |||
| indel_family_all += 1 | |||
| else: | |||
| pass | |||
| if mendelian == '1:1:1': | |||
| indel_family_mendelian += 1 | |||
| elif mendelian == 'Ref:1:1': | |||
| indel_family_mendelian += 1 | |||
| else: | |||
| pass | |||
| snv_sister = snv_sister_same/(snv_sister_same + snv_sister_diff) | |||
| indel_sister = indel_sister_same/(indel_sister_same + indel_sister_diff) | |||
| snv_quartet = snv_family_mendelian/snv_family_all | |||
| indel_quartet = indel_family_mendelian/indel_family_all | |||
| outcolumn = 'Family\tTotal_Variants\tMendelian_Concordant_Variants\tMendelian_Concordance_Rate\n' | |||
| indel_outResult = family + '.INDEL' + '\t' + str(indel_family_all) + '\t' + str(indel_family_mendelian) + '\t' + str(indel_quartet) + '\n' | |||
| snv_outResult = family + '.SNV' + '\t' + str(snv_family_all) + '\t' + str(snv_family_mendelian) + '\t' + str(snv_quartet) + '\n' | |||
| summary_file.write(outcolumn) | |||
| summary_file.write(indel_outResult) | |||
| summary_file.write(snv_outResult) | |||
+ 109
- 0
codescripts/oneClass.py
View File
| @@ -0,0 +1,109 @@ | |||
| # import modules | |||
| import numpy as np | |||
| import pandas as pd | |||
| from sklearn import svm | |||
| from sklearn import preprocessing | |||
| import sys, argparse, os | |||
| from vcf2bed import position_to_bed,padding_region | |||
| parser = argparse.ArgumentParser(description="this script is to preform one calss svm on each chromosome") | |||
| parser.add_argument('-train', '--trainDataset', type=str, help='training dataset generated from extracting vcf information part, with mutaitons supported by callsets', required=True) | |||
| parser.add_argument('-test', '--testDataset', type=str, help='testing dataset generated from extracting vcf information part, with mutaitons not called by all callsets', required=True) | |||
| parser.add_argument('-name', '--sampleName', type=str, help='sample name for output file name', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| train_input = args.trainDataset | |||
| test_input = args.testDataset | |||
| sample_name = args.sampleName | |||
| # default columns, which will be included in the included in the calssifier | |||
| chromosome = ['chr1','chr2','chr3','chr4','chr5','chr6','chr7','chr8','chr9','chr10','chr11','chr12','chr13','chr14','chr15' ,'chr16','chr17','chr18','chr19','chr20','chr21','chr22','chrX','chrY'] | |||
| feature_heter_cols = ['AltDP','BaseQRankSum','DB','DP','FS','GQ','MQ','MQRankSum','QD','ReadPosRankSum','RefDP','SOR','af'] | |||
| feature_homo_cols = ['AltDP','DB','DP','FS','GQ','MQ','QD','RefDP','SOR','af'] | |||
| # import datasets sepearate the records with or without BaseQRankSum annotation, etc. | |||
| def load_dat(dat_file_name): | |||
| dat = pd.read_table(dat_file_name) | |||
| dat['DB'] = dat['DB'].fillna(0) | |||
| dat = dat[dat['DP'] != 0] | |||
| dat['af'] = dat['AltDP']/(dat['AltDP'] + dat['RefDP']) | |||
| homo_rows = dat[dat['BaseQRankSum'].isnull()] | |||
| heter_rows = dat[dat['BaseQRankSum'].notnull()] | |||
| return homo_rows,heter_rows | |||
| train_homo,train_heter = load_dat(train_input) | |||
| test_homo,test_heter = load_dat(test_input) | |||
| clf = svm.OneClassSVM(nu=0.05,kernel='rbf', gamma='auto_deprecated',cache_size=500) | |||
| def prepare_dat(train_dat,test_dat,feature_cols,chromo): | |||
| chr_train = train_dat[train_dat['chromo'] == chromo] | |||
| chr_test = test_dat[test_dat['chromo'] == chromo] | |||
| train_dat = chr_train.loc[:,feature_cols] | |||
| test_dat = chr_test.loc[:,feature_cols] | |||
| train_dat_scaled = preprocessing.scale(train_dat) | |||
| test_dat_scaled = preprocessing.scale(test_dat) | |||
| return chr_test,train_dat_scaled,test_dat_scaled | |||
| def oneclass(X_train,X_test,chr_test): | |||
| clf.fit(X_train) | |||
| y_pred_test = clf.predict(X_test) | |||
| test_true_dat = chr_test[y_pred_test == 1] | |||
| test_false_dat = chr_test[y_pred_test == -1] | |||
| return test_true_dat,test_false_dat | |||
| predicted_true = pd.DataFrame(columns=train_homo.columns) | |||
| predicted_false = pd.DataFrame(columns=train_homo.columns) | |||
| for chromo in chromosome: | |||
| # homo datasets | |||
| chr_test_homo,X_train_homo,X_test_homo = prepare_dat(train_homo,test_homo,feature_homo_cols,chromo) | |||
| test_true_homo,test_false_homo = oneclass(X_train_homo,X_test_homo,chr_test_homo) | |||
| predicted_true = predicted_true.append(test_true_homo) | |||
| predicted_false = predicted_false.append(test_false_homo) | |||
| # heter datasets | |||
| chr_test_heter,X_train_heter,X_test_heter = prepare_dat(train_heter,test_heter,feature_heter_cols,chromo) | |||
| test_true_heter,test_false_heter = oneclass(X_train_heter,X_test_heter,chr_test_heter) | |||
| predicted_true = predicted_true.append(test_true_heter) | |||
| predicted_false = predicted_false.append(test_false_heter) | |||
| predicted_true_filename = sample_name + '_predicted_true.txt' | |||
| predicted_false_filename = sample_name + '_predicted_false.txt' | |||
| predicted_true.to_csv(predicted_true_filename,sep='\t',index=False) | |||
| predicted_false.to_csv(predicted_false_filename,sep='\t',index=False) | |||
| # output the bed file and padding bed region 50bp | |||
| predicted_true_bed_filename = sample_name + '_predicted_true.bed' | |||
| predicted_false_bed_filename = sample_name + '_predicted_false.bed' | |||
| padding_filename = sample_name + '_padding.bed' | |||
| predicted_true_bed = open(predicted_true_bed_filename,'w') | |||
| predicted_false_bed = open(predicted_false_bed_filename,'w') | |||
| padding = open(padding_filename,'w') | |||
| # | |||
| for index,row in predicted_false.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_false_bed.write(outline_pos) | |||
| chromo,pad_pos1,pad_pos2,pad_pos3,pad_pos4 = padding_region(chromo,pos1,pos2,50) | |||
| outline_pad_1 = chromo + '\t' + str(pad_pos1) + '\t' + str(pad_pos2) + '\n' | |||
| outline_pad_2 = chromo + '\t' + str(pad_pos3) + '\t' + str(pad_pos4) + '\n' | |||
| padding.write(outline_pad_1) | |||
| padding.write(outline_pad_2) | |||
| for index,row in predicted_true.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_true_bed.write(outline_pos) | |||
+ 101
- 0
codescripts/post-alignment.py
View File
| @@ -0,0 +1,101 @@ | |||
| import json | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import statistics | |||
| parser = argparse.ArgumentParser(description="This script is to summary information for pre-alignment QC") | |||
| parser.add_argument('-general', '--general_stat', type=str, help='multiqc_general_stats.txt', required=True) | |||
| parser.add_argument('-is', '--is_metrics', type=str, help='_is_metrics.txt', required=True) | |||
| parser.add_argument('-wgsmetrics', '--WgsMetricsAlgo', type=str, help='deduped_WgsMetricsAlgo', required=True) | |||
| parser.add_argument('-qualityyield', '--QualityYield', type=str, help='deduped_QualityYield', required=True) | |||
| parser.add_argument('-aln', '--aln_metrics', type=str, help='aln_metrics.txt', required=True) | |||
| args = parser.parse_args() | |||
| general_file = args.general_stat | |||
| is_file = args.is_metrics | |||
| wgsmetrics_file = args.wgsmetrics | |||
| qualityyield_file = args.qualityyield | |||
| aln_file = args.aln_metrics | |||
| ##### Table | |||
| ## general stat: % GC | |||
| dat = pd.read_table(general_file) | |||
| qualimap = dat.loc[:, dat.columns.str.startswith('QualiMap')] | |||
| qualimap.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| qualimap_stat = qualimap.dropna() | |||
| part1 = fastqc_stat.loc[:,['Sample', 'FastQC_mqc-generalstats-fastqc-percent_duplicates','FastQC_mqc-generalstats-fastqc-total_sequences']] | |||
| ## is_metrics: median insert size | |||
| ## deduped_WgsMetricsAlgo: 1x, 5x, 10x, 30x, median coverage | |||
| with open(html_file) as file: | |||
| origDict = json.load(file) | |||
| newdict = {(k1, k2):v2 for k1,v1 in origDict.items() \ | |||
| for k2,v2 in origDict[k1].items()} | |||
| df = pd.DataFrame([newdict[i] for i in sorted(newdict)], | |||
| index=pd.MultiIndex.from_tuples([i for i in sorted(newdict.keys())])) | |||
| gc = [] | |||
| at = [] | |||
| for i in part1['Sample']: | |||
| sub_df = df.loc[i,:] | |||
| gc.append(statistics.mean(sub_df['g']/sub_df['c'])) | |||
| at.append(statistics.mean(sub_df['a']/sub_df['t'])) | |||
| ## fastq_screen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| del fastqscreen['ERCC percentage'] | |||
| del fastqscreen['Phix percentage'] | |||
| ### merge all information | |||
| part1.insert(loc=3, column='G/C ratio', value=gc) | |||
| part1.insert(loc=4, column='A/T ratio', value=at) | |||
| part1.reset_index(drop=True, inplace=True) | |||
| fastqscreen.reset_index(drop=True, inplace=True) | |||
| df = pd.concat([part1, fastqscreen], axis=1) | |||
| df = df.append(df.mean(axis=0),ignore_index=True) | |||
| df = df.fillna('Batch average value') | |||
| df.columns = ['Sample','Total sequences (million)','% Dup','G/C ratio','A/T ratio','% Human','% EColi','% Adapter' , '% Vector','% rRNA' , '% Virus','% Yeast' ,'% Mitoch' ,'% No hits'] | |||
| df.to_csv('per-alignment_table_summary.txt',sep='\t',index=False) | |||
| ##### Picture | |||
| ## cumulative genome coverage | |||
| with open(json_file) as file: | |||
| all_dat = json.load(file) | |||
| genome_coverage_json = all_dat['report_plot_data']['qualimap_genome_fraction']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(genome_coverage_json) | |||
| genome_coverage = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| genome_coverage[dat.loc[i,'name']] = one_sample[1] | |||
| genome_coverage = genome_coverage.transpose() | |||
| genome_coverage['Sample'] = genome_coverage.index | |||
| genome_coverage.to_csv('post-alignment_genome_coverage.txt',sep='\t',index=False) | |||
| ## insert size histogram | |||
| insert_size_json = all_dat['report_plot_data']['qualimap_insert_size']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(insert_size_json) | |||
| insert_size = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| insert_size[dat.loc[i,'name']] = one_sample[1] | |||
| insert_size = insert_size.transpose() | |||
| insert_size['Sample'] = insert_size.index | |||
| insert_size.to_csv('post-alignment_insert_size.txt',sep='\t',index=False) | |||
| ## GC content distribution | |||
| gc_content_json = all_dat['report_plot_data']['qualimap_gc_content']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(gc_content_json) | |||
| gc_content = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| gc_content[dat.loc[i,'name']] = one_sample[1] | |||
| gc_content = gc_content.transpose() | |||
| gc_content['Sample'] = gc_content.index | |||
| gc_content.to_csv('post-alignment_gc_content.txt',sep='\t',index=False) | |||
+ 132
- 0
codescripts/pre_alignment.py
View File
| @@ -0,0 +1,132 @@ | |||
| import json | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import statistics | |||
| parser = argparse.ArgumentParser(description="This script is to summary information for pre-alignment QC") | |||
| parser.add_argument('-general', '--general_stat', type=str, help='multiqc_general_stats.txt', required=True) | |||
| parser.add_argument('-html', '--html', type=str, help='multiqc_report.html', required=True) | |||
| parser.add_argument('-fastqscreen', '--fastqscreen', type=str, help='multiqc_fastq_screen.txt', required=True) | |||
| parser.add_argument('-json', '--json', type=str, help='multiqc_happy_data.json', required=True) | |||
| args = parser.parse_args() | |||
| general_file = args.general_stat | |||
| html_file = args.html | |||
| fastqscreen_file = args.fastqscreen | |||
| json_file = args.json | |||
| ##### Table | |||
| ## general stat: 1. Total sequences; 2. %Dup | |||
| dat = pd.read_table(general_file) | |||
| fastqc = dat.loc[:, dat.columns.str.startswith('FastQC')] | |||
| fastqc.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_stat = fastqc.dropna() | |||
| part1 = fastqc_stat.loc[:,['Sample', 'FastQC_mqc-generalstats-fastqc-percent_duplicates','FastQC_mqc-generalstats-fastqc-total_sequences']] | |||
| ## report html: 1. G/C ratio; 2. A/T ratio | |||
| ## cat multiqc_report.html | grep 'fastqc_seq_content_data = ' | sed s'/fastqc_seq_content_data\ =\ //g' | sed 's/^[ \t]*//g' | sed s'/;//g' > fastqc_sequence_content.json | |||
| with open(html_file) as file: | |||
| origDict = json.load(file) | |||
| newdict = {(k1, k2):v2 for k1,v1 in origDict.items() \ | |||
| for k2,v2 in origDict[k1].items()} | |||
| df = pd.DataFrame([newdict[i] for i in sorted(newdict)], | |||
| index=pd.MultiIndex.from_tuples([i for i in sorted(newdict.keys())])) | |||
| gc = [] | |||
| at = [] | |||
| for i in part1['Sample']: | |||
| sub_df = df.loc[i,:] | |||
| gc.append(statistics.mean(sub_df['g']/sub_df['c'])) | |||
| at.append(statistics.mean(sub_df['a']/sub_df['t'])) | |||
| ## fastq_screen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| del fastqscreen['ERCC percentage'] | |||
| del fastqscreen['Phix percentage'] | |||
| ### merge all information | |||
| part1.insert(loc=3, column='G/C ratio', value=gc) | |||
| part1.insert(loc=4, column='A/T ratio', value=at) | |||
| part1.reset_index(drop=True, inplace=True) | |||
| fastqscreen.reset_index(drop=True, inplace=True) | |||
| df = pd.concat([part1, fastqscreen], axis=1) | |||
| df = df.append(df.mean(axis=0),ignore_index=True) | |||
| df = df.fillna('Batch average value') | |||
| df.columns = ['Sample','Total sequences (million)','% Dup','G/C ratio','A/T ratio','% Human','% EColi','% Adapter' , '% Vector','% rRNA' , '% Virus','% Yeast' ,'% Mitoch' ,'% No hits'] | |||
| df.to_csv('per-alignment_table_summary.txt',sep='\t',index=False) | |||
| ##### Picture | |||
| ## mean quality scores | |||
| with open(json_file) as file: | |||
| all_dat = json.load(file) | |||
| mean_quality_json = all_dat['report_plot_data']['fastqc_per_base_sequence_quality_plot']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(mean_quality_json) | |||
| mean_quality = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| mean_quality[dat.loc[i,'name']] = one_sample[1] | |||
| mean_quality = mean_quality.transpose() | |||
| mean_quality['Sample'] = mean_quality.index | |||
| mean_quality.to_csv('pre-alignment_mean_quality.txt',sep='\t',index=False) | |||
| ## per sequence GC content | |||
| gc_content_json = all_dat['report_plot_data']['fastqc_per_sequence_gc_content_plot']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(gc_content_json) | |||
| gc_content = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| gc_content[dat.loc[i,'name']] = one_sample[1] | |||
| gc_content = gc_content.transpose() | |||
| gc_content['Sample'] = gc_content.index | |||
| gc_content.to_csv('pre-alignment_gc_content.txt',sep='\t',index=False) | |||
| # fastqc and qualimap | |||
| dat = pd.read_table(fastqc_qualimap_file) | |||
| fastqc = dat.loc[:, dat.columns.str.startswith('FastQC')] | |||
| fastqc.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_stat = fastqc.dropna() | |||
| # qulimap | |||
| qualimap = dat.loc[:, dat.columns.str.startswith('QualiMap')] | |||
| qualimap.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| qualimap_stat = qualimap.dropna() | |||
| # fastqc | |||
| dat = pd.read_table(fastqc_file) | |||
| fastqc_module = dat.loc[:, "per_base_sequence_quality":"kmer_content"] | |||
| fastqc_module.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_all = pd.merge(fastqc_stat,fastqc_module, how='outer', left_on=['Sample'], right_on = ['Sample']) | |||
| # fastqscreen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| dat['Sample'] = [i.replace('_screen','') for i in dat['Sample']] | |||
| fastqscreen.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| # benchmark | |||
| with open(hap_file) as hap_json: | |||
| happy = json.load(hap_json) | |||
| dat =pd.DataFrame.from_records(happy) | |||
| dat = dat.loc[:, dat.columns.str.endswith('ALL')] | |||
| dat_transposed = dat.T | |||
| benchmark = dat_transposed.loc[:,['sample_id','METRIC.Precision','METRIC.Recall']] | |||
| benchmark['sample_id'] = benchmark.index | |||
| benchmark.columns = ['Sample','Precision','Recall'] | |||
| #output | |||
| fastqc_all.to_csv('fastqc.final.result.txt',sep="\t",index=0) | |||
| fastqscreen.to_csv('fastqscreen.final.result.txt',sep="\t",index=0) | |||
| qualimap_stat.to_csv('qualimap.final.result.txt',sep="\t",index=0) | |||
| benchmark.to_csv('benchmark.final.result.txt',sep="\t",index=0) | |||
+ 144
- 0
codescripts/reformVCF.py
View File
| @@ -0,0 +1,144 @@ | |||
| # import modules | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| parser = argparse.ArgumentParser(description="This script is to split samples in VCF files and rewrite to the right style") | |||
| parser.add_argument('-vcf', '--familyVCF', type=str, help='VCF with sister and mendelian infomation', required=True) | |||
| parser.add_argument('-name', '--familyName', type=str, help='Family name of the VCF file', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| inputFile = args.familyVCF | |||
| family_name = args.familyName | |||
| # output filename | |||
| LCL5_name = family_name + '.LCL5.vcf' | |||
| LCL5file = open(LCL5_name,'w') | |||
| LCL6_name = family_name + '.LCL6.vcf' | |||
| LCL6file = open(LCL6_name,'w') | |||
| LCL7_name = family_name + '.LCL7.vcf' | |||
| LCL7file = open(LCL7_name,'w') | |||
| LCL8_name = family_name + '.LCL8.vcf' | |||
| LCL8file = open(LCL8_name,'w') | |||
| family_filename = family_name + '.vcf' | |||
| familyfile = open(family_filename,'w') | |||
| # default columns, which will be included in the included in the calssifier | |||
| vcfheader = '''##fileformat=VCFv4.2 | |||
| ##FILTER=<ID=PASS,Description="the same genotype between twin sister and mendelian consistent in 578 and 678"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=0,Type=Flag,Description="0 for sister consistent, 1 for sister inconsistent"> | |||
| ##FORMAT=<ID=TRIO5,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##FORMAT=<ID=TRIO6,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # write VCF | |||
| LCL5colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL5'+'\n' | |||
| LCL5file.write(vcfheader) | |||
| LCL5file.write(LCL5colname) | |||
| LCL6colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL6'+'\n' | |||
| LCL6file.write(vcfheader) | |||
| LCL6file.write(LCL6colname) | |||
| LCL7colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL7'+'\n' | |||
| LCL7file.write(vcfheader) | |||
| LCL7file.write(LCL7colname) | |||
| LCL8colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL8'+'\n' | |||
| LCL8file.write(vcfheader) | |||
| LCL8file.write(LCL8colname) | |||
| familycolname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+'LCL5\t'+'LCL6\t'+'LCL7\t'+'LCL8'+'\n' | |||
| familyfile.write(vcfheader) | |||
| familyfile.write(familycolname) | |||
| # reform VCF | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| # replace . | |||
| # LCL6 uniq | |||
| if strings[11] == '.': | |||
| strings[11] = '0/0' | |||
| strings[9] = strings[12] | |||
| strings[10] = strings[13] | |||
| else: | |||
| pass | |||
| # LCL5 uniq | |||
| if strings[14] == '.': | |||
| strings[14] = '0/0' | |||
| strings[12] = strings[9] | |||
| strings[13] = strings[10] | |||
| else: | |||
| pass | |||
| # sister | |||
| if strings[11] == strings[14]: | |||
| add_format = ":1" | |||
| else: | |||
| add_format = ":0" | |||
| # trioLCL5 | |||
| if strings[15] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # trioLCL6 | |||
| if strings[7] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # filter | |||
| if (strings[11] == strings[14]) and (strings[15] == 'MD=1') and (strings[7] == 'MD=1'): | |||
| strings[6] = 'PASS' | |||
| else: | |||
| strings[6] = '.' | |||
| # output LCL5 | |||
| LCL5outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format + '\n' | |||
| LCL5file.write(LCL5outLine) | |||
| # output LCL6 | |||
| LCL6outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[11] + add_format + '\n' | |||
| LCL6file.write(LCL6outLine) | |||
| # output LCL7 | |||
| LCL7outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[10] + add_format + '\n' | |||
| LCL7file.write(LCL7outLine) | |||
| # output LCL8 | |||
| LCL8outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[9] + add_format + '\n' | |||
| LCL8file.write(LCL8outLine) | |||
| # output family | |||
| familyoutLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+ '.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format +'\t' + strings[11] + add_format + '\t' + strings[10] + add_format +'\t' + strings[9] + add_format + '\n' | |||
| familyfile.write(familyoutLine) | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| process(line) | |||
+ 227
- 0
codescripts/replicates_consensus.py
View File
| @@ -0,0 +1,227 @@ | |||
| from __future__ import division | |||
| from glob import glob | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='prefix of output file', required=True) | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| prefix = args.prefix | |||
| vcf = args.vcf | |||
| # input files | |||
| vcf_dat = pd.read_table(vcf) | |||
| # all info | |||
| all_file_name = prefix + "_all_summary.txt" | |||
| all_sample_outfile = open(all_file_name,'w') | |||
| all_info_col = 'CHROM\tPOS\tREF\tALT\tLCL5_consensus_calls\tLCL5_detect_number\tLCL5_same_diff\tLCL6_consensus_calls\tLCL6_detect_number\tLCl6_same_diff\tLCL7_consensus_calls\tLCL7_detect_number\tLCL7_same_diff\tLCL8_consensus_calls\tLCL8_detect_number\tLCL8_same_diff\n' | |||
| all_sample_outfile.write(all_info_col) | |||
| # filtered info | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200501 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| consensus_file_name = prefix + "_consensus.vcf" | |||
| consensus_outfile = open(consensus_file_name,'w') | |||
| consensus_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5_consensus_call\tLCL6_consensus_call\tLCL7_consensus_call\tLCL8_consensus_call\n' | |||
| consensus_outfile.write(vcf_header) | |||
| consensus_outfile.write(consensus_col) | |||
| # function | |||
| def decide_by_rep(vcf_list): | |||
| consensus_rep = '' | |||
| gt = [x.split(':')[0] for x in vcf_list] | |||
| gt_num_dict = Counter(gt) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if freq_gt >= 2: | |||
| consensus_rep = candidate_gt | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| return consensus_rep | |||
| def consensus_call(vcf_info_list): | |||
| consensus_call = '.' | |||
| detect_number = '.' | |||
| same_diff = '.' | |||
| # pcr | |||
| SEQ2000 = decide_by_rep(vcf_info_list[0:3]) | |||
| XTen_ARD = decide_by_rep(vcf_info_list[18:21]) | |||
| XTen_NVG = decide_by_rep(vcf_info_list[21:24]) | |||
| XTen_WUX = decide_by_rep(vcf_info_list[24:27]) | |||
| Nova_WUX = decide_by_rep(vcf_info_list[15:18]) | |||
| pcr_sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX,Nova_WUX] | |||
| pcr_sequence_dict = Counter(pcr_sequence_site) | |||
| pcr_highest_sequence = pcr_sequence_dict.most_common(1) | |||
| pcr_candidate_sequence = pcr_highest_sequence[0][0] | |||
| pcr_freq_sequence = pcr_highest_sequence[0][1] | |||
| if pcr_freq_sequence > 3: | |||
| pcr_consensus = pcr_candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| T7_WGE = decide_by_rep(vcf_info_list[3:6]) | |||
| Nova_ARD_1 = decide_by_rep(vcf_info_list[6:9]) | |||
| Nova_ARD_2 = decide_by_rep(vcf_info_list[9:12]) | |||
| Nova_BRG = decide_by_rep(vcf_info_list[12:15]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 2: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| gt = [x.replace('./.','.') for x in gt] | |||
| detected_num = 27 - gt.count('.') | |||
| gt_remain = [e for e in gt if e not in {'.'}] | |||
| gt_set = set(gt_remain) | |||
| if len(gt_set) == 1: | |||
| same_diff = 'same' | |||
| else: | |||
| same_diff = 'diff' | |||
| tag = ['inconGT','inconSequenceSite'] | |||
| if (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag): | |||
| consensus_call = pcr_consensus | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'notAgree' | |||
| else: | |||
| consensus_call = 'notConsensus' | |||
| return consensus_call, detected_num, same_diff | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| elif ((pcr_consensus == './.') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != './.')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| elif ((pcr_consensus != './.') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == './.')): | |||
| consensus_call = 'pcr-speicifc' | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = '0/0' | |||
| else: | |||
| consensus_call = 'filtered' | |||
| for row in vcf_dat.itertuples(): | |||
| # length | |||
| #alt | |||
| if ',' in row.ALT: | |||
| alt = row.ALT.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.ALT) | |||
| #ref | |||
| ref_len = len(row.REF) | |||
| if (alt_max > 50) or (ref_len > 50): | |||
| pass | |||
| else: | |||
| # consensus | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl6_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL6_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL6_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL6_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL6_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL6_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL6_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_3_20170216] | |||
| lcl7_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL7_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL7_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL7_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL7_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL7_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL7_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_3_20170216] | |||
| lcl8_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL8_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL8_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL8_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL8_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL8_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL8_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_3_20170216] | |||
| # LCL5 | |||
| LCL5_consensus_call, LCL5_detected_num, LCL5_same_diff = consensus_call(lcl5_list) | |||
| # LCL6 | |||
| LCL6_consensus_call, LCL6_detected_num, LCL6_same_diff = consensus_call(lcl6_list) | |||
| # LCL7 | |||
| LCL7_consensus_call, LCL7_detected_num, LCL7_same_diff = consensus_call(lcl7_list) | |||
| # LCL8 | |||
| LCL8_consensus_call, LCL8_detected_num, LCL8_same_diff = consensus_call(lcl8_list) | |||
| # all data | |||
| all_output = row._1 + '\t' + str(row.POS) + '\t' + row.REF + '\t' + row.ALT + '\t' + LCL5_consensus_call + '\t' + str(LCL5_detected_num) + '\t' + LCL5_same_diff + '\t' +\ | |||
| LCL6_consensus_call + '\t' + str(LCL6_detected_num) + '\t' + LCL6_same_diff + '\t' +\ | |||
| LCL7_consensus_call + '\t' + str(LCL7_detected_num) + '\t' + LCL7_same_diff + '\t' +\ | |||
| LCL8_consensus_call + '\t' + str(LCL8_detected_num) + '\t' + LCL8_same_diff + '\n' | |||
| all_sample_outfile.write(all_output) | |||
| #consensus vcf | |||
| one_position = [LCL5_consensus_call,LCL6_consensus_call,LCL7_consensus_call,LCL8_consensus_call] | |||
| if ('notConsensus' in one_position) or (((len(set(one_position)) == 1) and ('./.' in set(one_position))) or ((len(set(one_position)) == 1) and ('0/0' in set(one_position))) or ((len(set(one_position)) == 2) and ('0/0' in set(one_position) and ('./.' in set(one_position))))): | |||
| pass | |||
| else: | |||
| consensus_output = row._1 + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + row.ALT + '\t' + '.' + '\t' + '.' + '\t' +'.' + '\t' + 'GT' + '\t' + LCL5_consensus_call + '\t' + LCL6_consensus_call + '\t' + LCL7_consensus_call + '\t' + LCL8_consensus_call +'\n' | |||
| consensus_outfile.write(consensus_output) | |||
+ 36
- 0
codescripts/vcf2bed.py
View File
| @@ -0,0 +1,36 @@ | |||
| import re | |||
| def position_to_bed(chromo,pos,ref,alt): | |||
| # snv | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF | |||
| if len(ref) == 1 and len(alt) == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| # deletions | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (reference length - alternate length) | |||
| elif len(ref) > 1 and len(alt) == 1: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| #insertions | |||
| # For insertions: | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (alternate length - reference length) | |||
| else: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(alt) - 1) | |||
| return chromo,StartPos,EndPos | |||
| def padding_region(chromo,pos1,pos2,padding): | |||
| StartPos1 = pos1 - padding | |||
| EndPos1 = pos1 | |||
| StartPos2 = pos2 | |||
| EndPos2 = pos2 + padding | |||
| return chromo,StartPos1,EndPos1,StartPos2,EndPos2 | |||
+ 295
- 0
codescripts/voted_by_vcfinfo_mendelianinfo.py
View File
| @@ -0,0 +1,295 @@ | |||
| from __future__ import division | |||
| from glob import glob | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-folder', '--folder', type=str, help='directory that holds all the mendelian info', required=True) | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| folder = args.folder | |||
| vcf = args.vcf | |||
| # input files | |||
| folder = folder + '/*.txt' | |||
| filenames = glob(folder) | |||
| dataframes = [] | |||
| for filename in filenames: | |||
| dataframes.append(pd.read_table(filename,header=None)) | |||
| dfs = [df.set_index([0, 1]) for df in dataframes] | |||
| merged_mendelian = pd.concat(dfs, axis=1).reset_index() | |||
| family_name = [i.split('/')[-1].replace('.txt','') for i in filenames] | |||
| columns = ['CHROM','POS'] + family_name | |||
| merged_mendelian.columns = columns | |||
| vcf_dat = pd.read_table(vcf) | |||
| merged_df = pd.merge(merged_mendelian, vcf_dat, how='outer', left_on=['CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_df = merged_df.fillna('nan') | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200501 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Sum depth of all samples"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Sum alternative depth of all samples"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # output files | |||
| benchmark_LCL5 = open('LCL5_voted.vcf','w') | |||
| benchmark_LCL6 = open('LCL6_voted.vcf','w') | |||
| benchmark_LCL7 = open('LCL7_voted.vcf','w') | |||
| benchmark_LCL8 = open('LCL8_voted.vcf','w') | |||
| all_sample_outfile = open('all_sample_information.txt','w') | |||
| # write VCF | |||
| LCL5_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5_benchmark_calls\n' | |||
| LCL6_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL6_benchmark_calls\n' | |||
| LCL7_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL7_benchmark_calls\n' | |||
| LCL8_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL8_benchmark_calls\n' | |||
| benchmark_LCL5.write(vcf_header) | |||
| benchmark_LCL5.write(LCL5_col) | |||
| benchmark_LCL6.write(vcf_header) | |||
| benchmark_LCL6.write(LCL6_col) | |||
| benchmark_LCL7.write(vcf_header) | |||
| benchmark_LCL7.write(LCL7_col) | |||
| benchmark_LCL8.write(vcf_header) | |||
| benchmark_LCL8.write(LCL8_col) | |||
| # all info | |||
| all_info_col = 'CHROM\tPOS\tLCL5_pcr_consensus\tLCL5_pcr_free_consensus\tLCL5_mendelian_num\tLCL5_consensus_call\tLCL5_consensus_alt_seq\tLCL5_alt\tLCL5_dp\tLCL5_detected_num\tLCL6_pcr_consensus\tLCL6_pcr_free_consensus\tLCL6_mendelian_num\tLCL6_consensus_call\tLCL6_consensus_alt_seq\tLCL6_alt\tLCL6_dp\tLCL6_detected_num\tLCL7_pcr_consensus\tLCL7_pcr_free_consensus\tLCL7_mendelian_num\t LCL7_consensus_call\tLCL7_consensus_alt_seq\tLCL7_alt\tLCL7_dp\tLCL7_detected_num\tLCL8_pcr_consensus\tLCL8_pcr_free_consensus\tLCL8_mendelian_num\tLCL8_consensus_call\tLCL8_consensus_alt_seq\tLCL8_alt\tLCL8_dp\tLCL8_detected_num\n' | |||
| all_sample_outfile.write(all_info_col) | |||
| # function | |||
| def decide_by_rep(vcf_list,mendelian_list): | |||
| consensus_rep = '' | |||
| gt = [x.split(':')[0] for x in vcf_list] | |||
| # mendelian consistent? | |||
| mendelian_dict = Counter(mendelian_list) | |||
| highest_mendelian = mendelian_dict.most_common(1) | |||
| candidate_mendelian = highest_mendelian[0][0] | |||
| freq_mendelian = highest_mendelian[0][1] | |||
| if (candidate_mendelian == '1:1:1') and (freq_mendelian >= 2): | |||
| con_loc = [i for i in range(len(mendelian_list)) if mendelian_list[i] == '1:1:1'] | |||
| gt_con = itemgetter(*con_loc)(gt) | |||
| gt_num_dict = Counter(gt_con) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if (candidate_gt != './.') and (freq_gt >= 2): | |||
| consensus_rep = candidate_gt | |||
| elif (candidate_gt == './.') and (freq_gt >= 2): | |||
| consensus_rep = 'noGTInfo' | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| elif (candidate_mendelian == 'nan') and (freq_mendelian >= 2): | |||
| consensus_rep = 'noMenInfo' | |||
| else: | |||
| consensus_rep = 'inconMen' | |||
| return consensus_rep | |||
| def consensus_call(vcf_info_list,mendelian_list,alt_seq): | |||
| pcr_consensus = '.' | |||
| pcr_free_consensus = '.' | |||
| mendelian_num = '.' | |||
| consensus_call = '.' | |||
| consensus_alt_seq = '.' | |||
| # pcr | |||
| SEQ2000 = decide_by_rep(vcf_info_list[0:3],mendelian_list[0:3]) | |||
| XTen_ARD = decide_by_rep(vcf_info_list[18:21],mendelian_list[18:21]) | |||
| XTen_NVG = decide_by_rep(vcf_info_list[21:24],mendelian_list[21:24]) | |||
| XTen_WUX = decide_by_rep(vcf_info_list[24:27],mendelian_list[24:27]) | |||
| pcr_sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX] | |||
| pcr_sequence_dict = Counter(pcr_sequence_site) | |||
| pcr_highest_sequence = pcr_sequence_dict.most_common(1) | |||
| pcr_candidate_sequence = pcr_highest_sequence[0][0] | |||
| pcr_freq_sequence = pcr_highest_sequence[0][1] | |||
| if pcr_freq_sequence > 2: | |||
| pcr_consensus = pcr_candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| T7_WGE = decide_by_rep(vcf_info_list[3:6],mendelian_list[3:6]) | |||
| Nova_ARD_1 = decide_by_rep(vcf_info_list[6:9],mendelian_list[6:9]) | |||
| Nova_ARD_2 = decide_by_rep(vcf_info_list[9:12],mendelian_list[9:12]) | |||
| Nova_BRG = decide_by_rep(vcf_info_list[12:15],mendelian_list[12:15]) | |||
| Nova_WUX = decide_by_rep(vcf_info_list[15:18],mendelian_list[15:18]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG,Nova_WUX] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 3: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| # net alt, dp | |||
| # alt | |||
| AD = [x.split(':')[1] for x in vcf_info_list] | |||
| ALT = [x.split(',')[1] for x in AD] | |||
| ALT = [int(x) for x in ALT] | |||
| ALL_ALT = sum(ALT) | |||
| # dp | |||
| DP = [x.split(':')[2] for x in vcf_info_list] | |||
| DP = [int(x) for x in DP] | |||
| ALL_DP = sum(DP) | |||
| # detected number | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| gt = [x.replace('0/0','.') for x in gt] | |||
| gt = [x.replace('./.','.') for x in gt] | |||
| detected_num = 27 - gt.count('.') | |||
| # decide consensus calls | |||
| tag = ['inconGT','noMenInfo','inconMen','inconSequenceSite','noGTInfo'] | |||
| if (pcr_consensus != '0/0') and (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag): | |||
| consensus_call = pcr_consensus | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| indices = [i for i, x in enumerate(gt) if x == consensus_call] | |||
| matched_mendelian = itemgetter(*indices)(mendelian_list) | |||
| mendelian_num = matched_mendelian.count('1:1:1') | |||
| # Delete multiple alternative genotype to necessary expression | |||
| alt_gt = alt_seq.split(',') | |||
| if len(alt_gt) > 1: | |||
| allele1 = consensus_call.split('/')[0] | |||
| allele2 = consensus_call.split('/')[1] | |||
| if allele1 == '0': | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| consensus_alt_seq = allele2_seq | |||
| consensus_call = '0/1' | |||
| else: | |||
| allele1_seq = alt_gt[int(allele1) - 1] | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| if int(allele1) > int(allele2): | |||
| consensus_alt_seq = allele2_seq + ',' + allele1_seq | |||
| consensus_call = '1/2' | |||
| elif int(allele1) < int(allele2): | |||
| consensus_alt_seq = allele1_seq + ',' + allele2_seq | |||
| consensus_call = '1/2' | |||
| else: | |||
| consensus_alt_seq = allele1_seq | |||
| consensus_call = '1/1' | |||
| else: | |||
| consensus_alt_seq = alt_seq | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| elif ((pcr_consensus == './.') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != './.')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| elif ((pcr_consensus != './.') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == './.')): | |||
| consensus_call = 'pcr-speicifc' | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = '0/0' | |||
| else: | |||
| consensus_call = 'filtered' | |||
| return pcr_consensus, pcr_free_consensus, mendelian_num, consensus_call, consensus_alt_seq, ALL_ALT, ALL_DP, detected_num | |||
| for row in merged_df.itertuples(): | |||
| mendelian_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_1_20191105,row.Quartet_DNA_BGI_T7_WGE_2_20191105,row.Quartet_DNA_BGI_T7_WGE_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_3_20170216] | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl6_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL6_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL6_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL6_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL6_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL6_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL6_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_3_20170216] | |||
| lcl7_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL7_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL7_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL7_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL7_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL7_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL7_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_3_20170216] | |||
| lcl8_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL8_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL8_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL8_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL8_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL8_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL8_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_3_20170216] | |||
| # LCL5 | |||
| LCL5_pcr_consensus, LCL5_pcr_free_consensus, LCL5_mendelian_num, LCL5_consensus_call, LCL5_consensus_alt_seq, LCL5_alt, LCL5_dp, LCL5_detected_num = consensus_call(lcl5_list,mendelian_list,row.ALT) | |||
| if LCL5_mendelian_num != '.': | |||
| LCL5_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL5_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL5_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL5_consensus_call + ':' + str(LCL5_alt) + ':' + str(LCL5_dp) + '\n' | |||
| benchmark_LCL5.write(LCL5_output) | |||
| # LCL6 | |||
| LCL6_pcr_consensus, LCL6_pcr_free_consensus, LCL6_mendelian_num, LCL6_consensus_call, LCL6_consensus_alt_seq, LCL6_alt, LCL6_dp, LCL6_detected_num = consensus_call(lcl6_list,mendelian_list,row.ALT) | |||
| if LCL6_mendelian_num != '.': | |||
| LCL6_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL6_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL6_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL6_consensus_call + ':' + str(LCL6_alt) + ':' + str(LCL6_dp) + '\n' | |||
| benchmark_LCL6.write(LCL6_output) | |||
| # LCL7 | |||
| LCL7_pcr_consensus, LCL7_pcr_free_consensus, LCL7_mendelian_num, LCL7_consensus_call, LCL7_consensus_alt_seq, LCL7_alt, LCL7_dp, LCL7_detected_num = consensus_call(lcl7_list,mendelian_list,row.ALT) | |||
| if LCL7_mendelian_num != '.': | |||
| LCL7_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL7_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL7_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL7_consensus_call + ':' + str(LCL7_alt) + ':' + str(LCL7_dp) + '\n' | |||
| benchmark_LCL7.write(LCL7_output) | |||
| # LCL8 | |||
| LCL8_pcr_consensus, LCL8_pcr_free_consensus, LCL8_mendelian_num, LCL8_consensus_call, LCL8_consensus_alt_seq, LCL8_alt, LCL8_dp, LCL8_detected_num = consensus_call(lcl8_list,mendelian_list,row.ALT) | |||
| if LCL8_mendelian_num != '.': | |||
| LCL8_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL8_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL8_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL8_consensus_call + ':' + str(LCL8_alt) + ':' + str(LCL8_dp) + '\n' | |||
| benchmark_LCL8.write(LCL8_output) | |||
| # all data | |||
| all_output = row.CHROM + '\t' + str(row.POS) + '\t' + LCL5_pcr_consensus + '\t' + LCL5_pcr_free_consensus + '\t' + str(LCL5_mendelian_num) + '\t' + LCL5_consensus_call + '\t' + LCL5_consensus_alt_seq + '\t' + str(LCL5_alt) + '\t' + str(LCL5_dp) + '\t' + str(LCL5_detected_num) + '\t' +\ | |||
| LCL6_pcr_consensus + '\t' + LCL6_pcr_free_consensus + '\t' + str(LCL6_mendelian_num) + '\t' + LCL6_consensus_call + '\t' + LCL6_consensus_alt_seq + '\t' + str(LCL6_alt) + '\t' + str(LCL6_dp) + '\t' + str(LCL6_detected_num) + '\t' +\ | |||
| LCL7_pcr_consensus + '\t' + LCL7_pcr_free_consensus + '\t' + str(LCL7_mendelian_num) + '\t' + LCL7_consensus_call + '\t' + LCL7_consensus_alt_seq + '\t' + str(LCL7_alt) + '\t' + str(LCL7_dp) + '\t' + str(LCL7_detected_num) + '\t' +\ | |||
| LCL8_pcr_consensus + '\t' + LCL8_pcr_free_consensus + '\t' + str(LCL8_mendelian_num) + '\t' + LCL8_consensus_call + '\t' + LCL8_consensus_alt_seq + '\t' + str(LCL8_alt) + '\t' + str(LCL8_dp) + '\t' + str(LCL8_detected_num) + '\n' | |||
| all_sample_outfile.write(all_output) | |||
+ 28
- 0
defaults
View File
| @@ -0,0 +1,28 @@ | |||
| { | |||
| "benchmarking_dir": "oss://pgx-result/renluyao/manuscript_v3.0/reference_datasets_v202103/", | |||
| "SENTIEON_INSTALL_DIR": "/opt/sentieon-genomics", | |||
| "fasta": "GRCh38.d1.vd1.fa", | |||
| "BENCHMARKdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-hap:latest", | |||
| "dbsnp_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "BEDTOOLSdocker": "registry-internal.cn-shanghai.aliyuncs.com/pgx-docker-registry/bedtools:v2.27.1", | |||
| "disk_size": "500", | |||
| "FASTQCdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqc:0.11.8", | |||
| "MULTIQCdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/multiqc:v1.8", | |||
| "SMALLcluster_config": "OnDemand bcs.ps.g.xlarge img-ubuntu-vpc", | |||
| "screen_ref_dir": "oss://pgx-reference-data/fastq_screen_reference/", | |||
| "benchmark_region": "oss://pgx-result/renluyao/manuscript_v3.0/reference_datasets_v202103/Quartet.high.confidence.region.v202103.bed", | |||
| "dbmills_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "BIGcluster_config": "OnDemand bcs.a2.7xlarge img-ubuntu-vpc", | |||
| "fastq_screen_conf": "oss://pgx-reference-data/fastq_screen_reference/fastq_screen.conf", | |||
| "FASTQSCREENdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqscreen:0.12.0", | |||
| "SENTIEON_LICENSE": "192.168.0.55:8990", | |||
| "SENTIEONdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/sentieon-genomics:v2019.11.28", | |||
| "QUALIMAPdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/qualimap:2.0.0", | |||
| "db_mills": "Mills_and_1000G_gold_standard.indels.hg38.vcf", | |||
| "dbsnp": "dbsnp_146.hg38.vcf", | |||
| "MENDELIANdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/vbt:v1.1", | |||
| "DIYdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.4", | |||
| "ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/" | |||
| } | |||
+ 4
- 0
inputSamples.Example.txt
View File
| @@ -0,0 +1,4 @@ | |||
| oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL5_1_20171025_R1.fastq.gz oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL5_1_20171025_R2.fastq.gz Quartet_DNA_ILM_Nova_GAC_LCL5_1_20171025 | |||
| oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL6_1_20171025_R1.fastq.gz oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL6_1_20171025_R2.fastq.gz Quartet_DNA_ILM_Nova_GAC_LCL6_1_20171025 | |||
| oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL7_1_20171025_R1.fastq.gz oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL7_1_20171025_R2.fastq.gz Quartet_DNA_ILM_Nova_GAC_LCL7_1_20171025 | |||
| oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL8_1_20171025_R1.fastq.gz oss://chinese-quartet/quartet-storage-data/Short_reads_fastq/ILM_Nova_GAC_DNA/Quartet_DNA_ILM_Nova_GAC_LCL8_1_20171025_R2.fastq.gz Quartet_DNA_ILM_Nova_GAC_LCL8_1_20171025 | |||
+ 29
- 0
inputs
View File
| @@ -0,0 +1,29 @@ | |||
| { | |||
| "{{ project_name }}.benchmarking_dir": "{{ benchmarking_dir }}", | |||
| "{{ project_name }}.SENTIEON_INSTALL_DIR": "{{ SENTIEON_INSTALL_DIR }}", | |||
| "{{ project_name }}.fasta": "{{ fasta }}", | |||
| "{{ project_name }}.BENCHMARKdocker": "{{ BENCHMARKdocker }}", | |||
| "{{ project_name }}.dbsnp_dir": "{{ dbsnp_dir }}", | |||
| "{{ project_name }}.BEDTOOLSdocker": "{{ BEDTOOLSdocker }}", | |||
| "{{ project_name }}.disk_size": "{{ disk_size }}", | |||
| "{{ project_name }}.inputSamplesFile": "{{ inputSamplesFile }}", | |||
| "{{ project_name }}.FASTQCdocker": "{{ FASTQCdocker }}", | |||
| "{{ project_name }}.MULTIQCdocker": "{{ MULTIQCdocker }}", | |||
| "{{ project_name }}.project": "{{ project }}", | |||
| "{{ project_name }}.SMALLcluster_config": "{{ SMALLcluster_config }}", | |||
| "{{ project_name }}.screen_ref_dir": "{{ screen_ref_dir }}", | |||
| "{{ project_name }}.bed": "{{ bed }}", | |||
| "{{ project_name }}.dbmills_dir": "{{ dbmills_dir }}", | |||
| "{{ project_name }}.BIGcluster_config": "{{ BIGcluster_config }}", | |||
| "{{ project_name }}.fastq_screen_conf": "{{ fastq_screen_conf }}", | |||
| "{{ project_name }}.FASTQSCREENdocker": "{{ FASTQSCREENdocker }}", | |||
| "{{ project_name }}.SENTIEON_LICENSE": "{{ SENTIEON_LICENSE }}", | |||
| "{{ project_name }}.SENTIEONdocker": "{{ SENTIEONdocker }}", | |||
| "{{ project_name }}.QUALIMAPdocker": "{{ QUALIMAPdocker }}", | |||
| "{{ project_name }}.benchmark_region": "{{ benchmark_region }}", | |||
| "{{ project_name }}.db_mills": "{{ db_mills }}", | |||
| "{{ project_name }}.dbsnp": "{{ dbsnp }}", | |||
| "{{ project_name }}.MENDELIANdocker": "{{ MENDELIANdocker }}", | |||
| "{{ project_name }}.DIYdocker": "{{ DIYdocker }}", | |||
| "{{ project_name }}.ref_dir": "{{ ref_dir }}" | |||
| } | |||
BIN
pictures/Picture1.png
View File
{kind=link}
| Before | After |
|---|---|
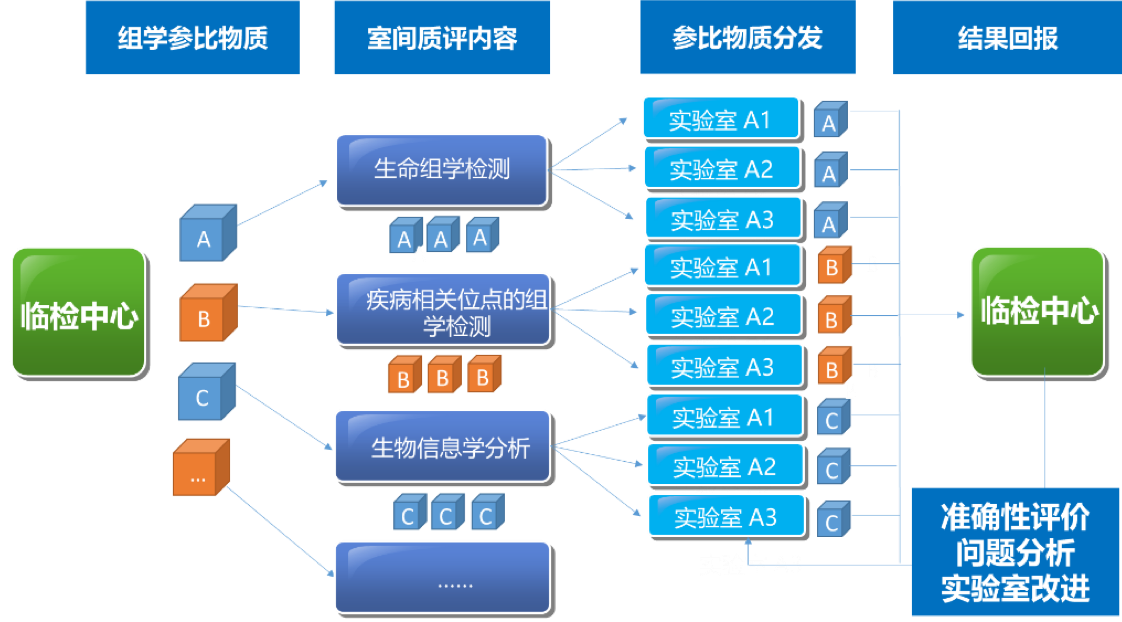
|
|
| Width: 1122 | Height: 622 | Size: 240KB |
BIN
pictures/performance.png
View File
{kind=link}
| Before | After |
|---|---|
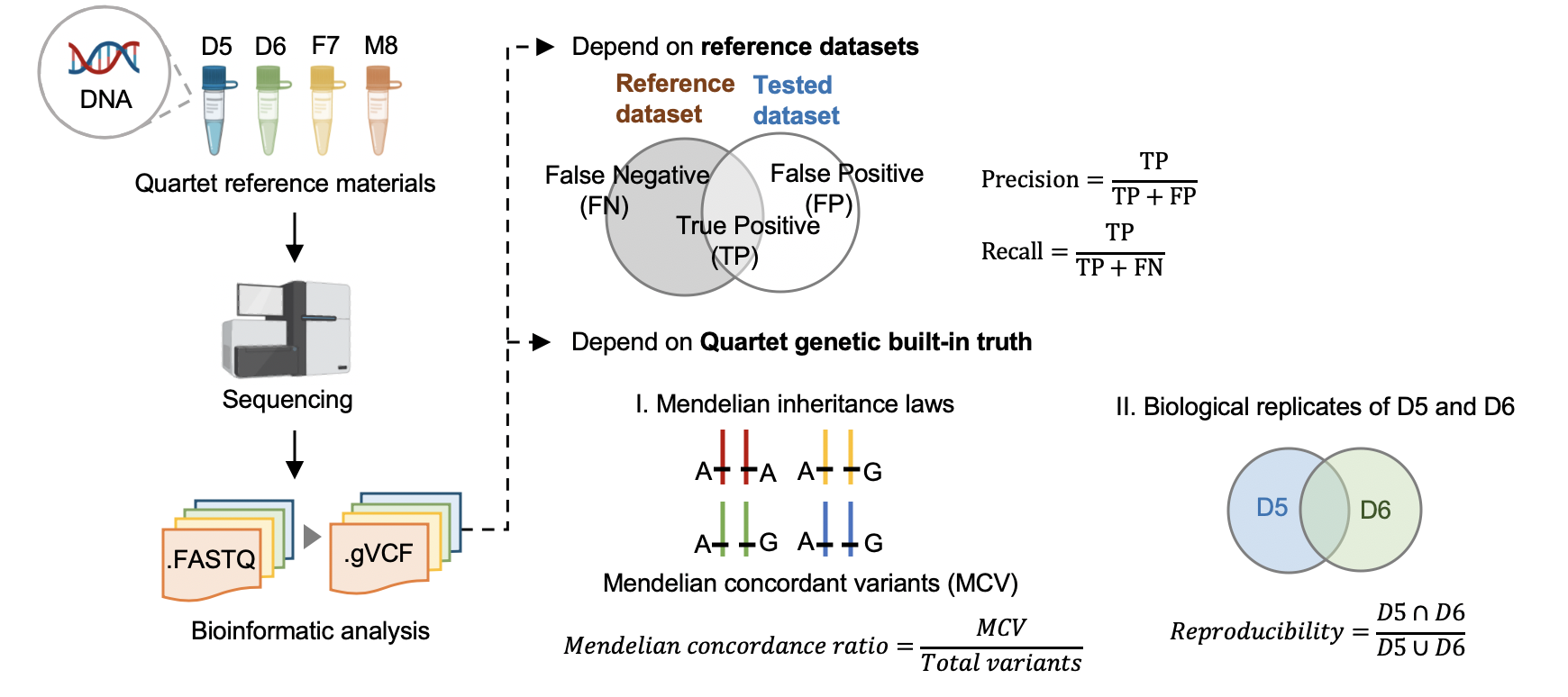
|
|
| Width: 1740 | Height: 774 | Size: 387KB |
BIN
pictures/table.png
View File
{kind=link}
| Before | After |
|---|---|
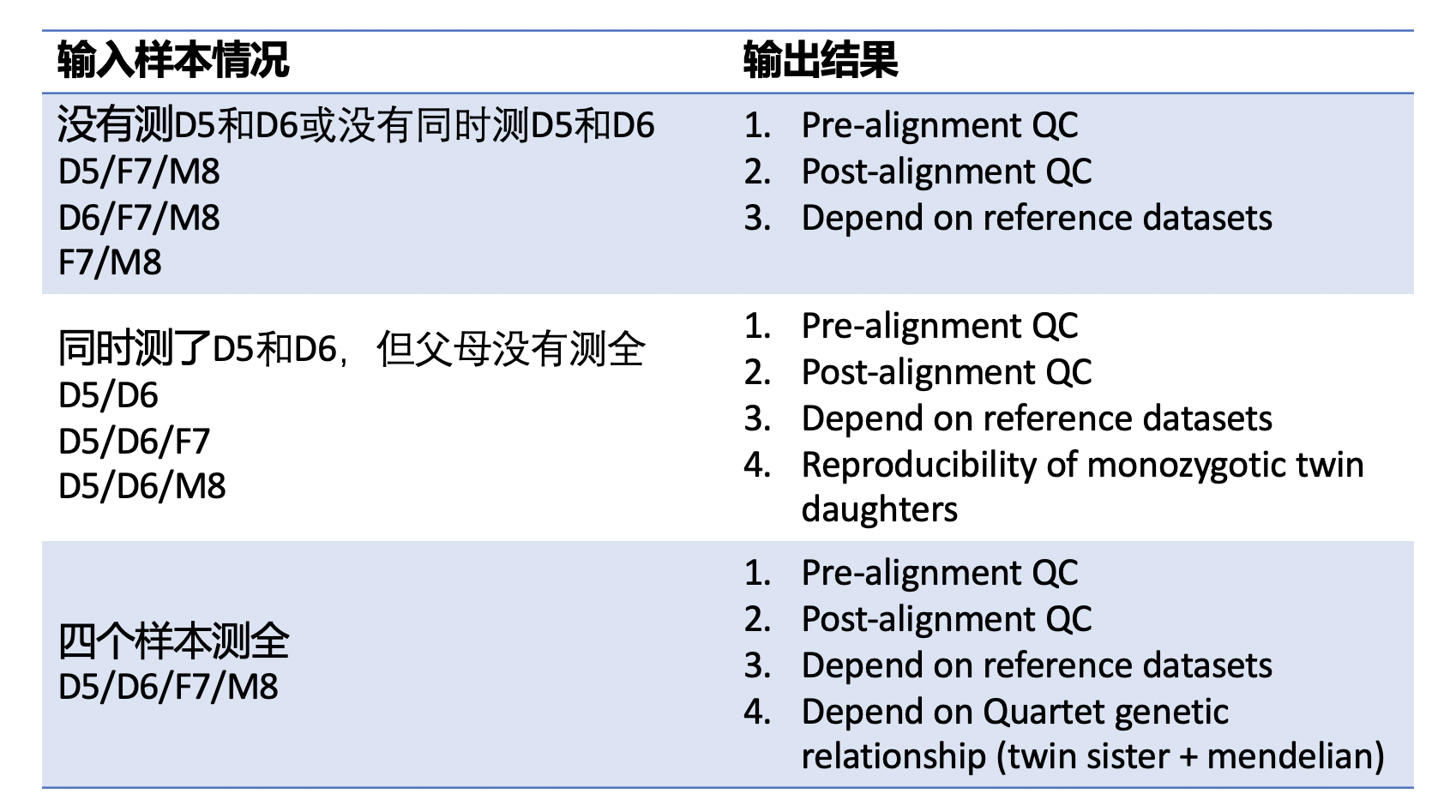
|
|
| Width: 1698 | Height: 947 | Size: 448KB |
BIN
pictures/workflow.png
View File
{kind=link}
| Before | After |
|---|---|
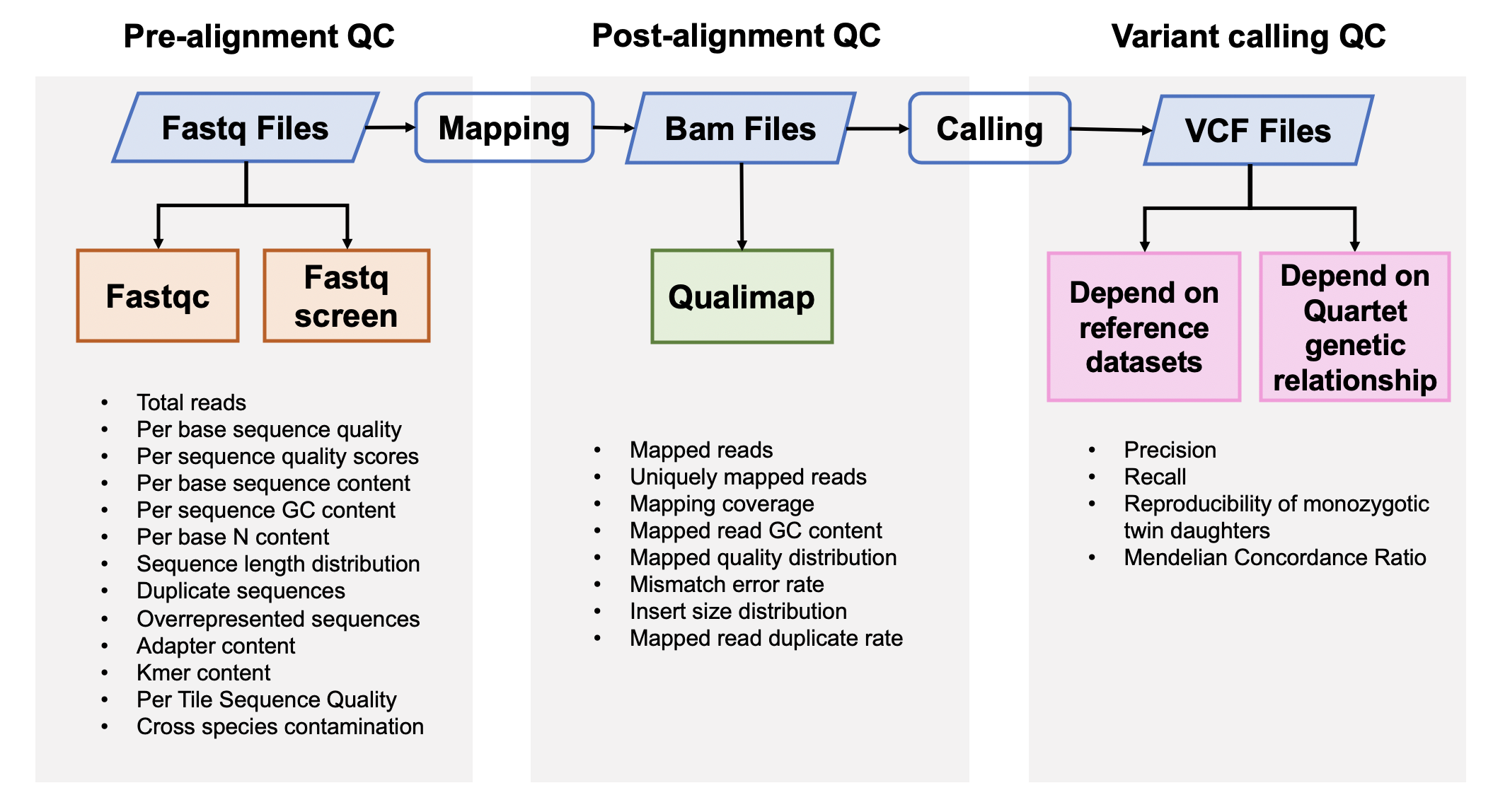
|
|
| Width: 2110 | Height: 1148 | Size: 599KB |
BIN
tasks/.DS_Store
View File
+ 48
- 0
tasks/BQSR.wdl
View File
| @@ -0,0 +1,48 @@ | |||
| task BQSR { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File realigned_bam | |||
| File realigned_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} -q ${sample}_recal_data.table --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table.post --algo ReadWriter ${sample}.sorted.deduped.realigned.recaled.bam | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt --algo QualCal --plot --before ${sample}_recal_data.table --after ${sample}_recal_data.table.post ${sample}_recal_data.csv | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot QualCal -o ${sample}_bqsrreport.pdf ${sample}_recal_data.csv | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File recal_table = "${sample}_recal_data.table" | |||
| File recal_post = "${sample}_recal_data.table.post" | |||
| File recaled_bam = "${sample}.sorted.deduped.realigned.recaled.bam" | |||
| File recaled_bam_index = "${sample}.sorted.deduped.realigned.recaled.bam.bai" | |||
| File recal_csv = "${sample}_recal_data.csv" | |||
| File bqsrreport_pdf = "${sample}_bqsrreport.pdf" | |||
| } | |||
| } | |||
+ 49
- 0
tasks/D5_D6.wdl
View File
| @@ -0,0 +1,49 @@ | |||
| task D5_D6 { | |||
| Array[File] splited_vcf | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| mkdir -p /cromwell_root/tmp/vcf | |||
| cp ${sep=" " splited_vcf} /cromwell_root/tmp/vcf | |||
| for i in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| for j in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| sample_i=$(echo $i | cut -f7 -d_) | |||
| sample_j=$(echo $j | cut -f7 -d_) | |||
| rep_i=$(echo $i | cut -f8 -d_) | |||
| rep_j=$(echo $j | cut -f8 -d_) | |||
| if [ $sample_i == "LCL5" ] && [ $sample_j == "LCL6" ] && [ $rep_i == $rep_j ];then | |||
| cat $i | grep -v '##' | cut -f1,2,4,5,10 | cut -d ':' -f1 > LCL5.txt | |||
| cat $j | grep -v '##' | cut -f10 | cut -d ':' -f1 > LCL6.txt | |||
| paste LCL5.txt LCL6.txt > sister.txt | |||
| python /opt/D5_D6.py -sister sister.txt -project ${project}.$rep_i | |||
| fi | |||
| done | |||
| done | |||
| for i in *.reproducibility.txt | |||
| do | |||
| cat $i | sed -n '2,2p' >> sister.summary | |||
| done | |||
| sed '1i\Family\tReproducibility_D5_D6' sister.summary > ${project}.sister.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| Array[File] sister_file = glob("*.reproducibility.txt") | |||
| File sister_summary = "${project}.sister.txt" | |||
| } | |||
| } | |||
+ 40
- 0
tasks/Dedup.wdl
View File
| @@ -0,0 +1,40 @@ | |||
| task Dedup { | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo LocusCollector --fun score_info ${sample}_score.txt | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo Dedup --rmdup --score_info ${sample}_score.txt --metrics ${sample}_dedup_metrics.txt ${sample}.sorted.deduped.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File score = "${sample}_score.txt" | |||
| File dedup_metrics = "${sample}_dedup_metrics.txt" | |||
| File Dedup_bam = "${sample}.sorted.deduped.bam" | |||
| File Dedup_bam_index = "${sample}.sorted.deduped.bam.bai" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/GVCFtyper.wdl
View File
| @@ -0,0 +1,34 @@ | |||
| task GVCFtyper { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| Array[File] vcf | |||
| Array[File] vcf_idx | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} --algo GVCFtyper ${project}.joint.g.vcf ${sep=" " vcf} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File gvcf = "${project}.joint.g.vcf" | |||
| File gvcf_idx = "${project}.joint.g.vcf.idx" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/Haplotyper_gVCF.wdl
View File
| @@ -0,0 +1,34 @@ | |||
| task Haplotyper_gVCF { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File recaled_bam | |||
| File recaled_bam_index | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper --emit_mode gvcf ${sample}_hc.g.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf = "${sample}_hc.g.vcf" | |||
| File vcf_idx = "${sample}_hc.g.vcf.idx" | |||
| } | |||
| } | |||
+ 56
- 0
tasks/Metrics.wdl
View File
| @@ -0,0 +1,56 @@ | |||
| task Metrics { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${sorted_bam} --algo MeanQualityByCycle ${sample}_mq_metrics.txt --algo QualDistribution ${sample}_qd_metrics.txt --algo GCBias --summary ${sample}_gc_summary.txt ${sample}_gc_metrics.txt --algo AlignmentStat ${sample}_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_is_metrics.txt --algo CoverageMetrics --omit_base_output ${sample}_coverage_metrics | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot metrics -o ${sample}_metrics_report.pdf gc=${sample}_gc_metrics.txt qd=${sample}_qd_metrics.txt mq=${sample}_mq_metrics.txt isize=${sample}_is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File qd_metrics = "${sample}_qd_metrics.txt" | |||
| File qd_metrics_pdf = "${sample}_qd_metrics.pdf" | |||
| File mq_metrics = "${sample}_mq_metrics.txt" | |||
| File mq_metrics_pdf = "${sample}_mq_metrics.pdf" | |||
| File is_metrics = "${sample}_is_metrics.txt" | |||
| File is_metrics_pdf = "${sample}_is_metrics.pdf" | |||
| File gc_summary = "${sample}_gc_summary.txt" | |||
| File gc_metrics = "${sample}_gc_metrics.txt" | |||
| File gc_metrics_pdf = "${sample}_gc_metrics.pdf" | |||
| File aln_metrics = "${sample}_aln_metrics.txt" | |||
| File coverage_metrics_sample_summary = "${sample}_coverage_metrics.sample_summary" | |||
| File coverage_metrics_sample_statistics = "${sample}_coverage_metrics.sample_statistics" | |||
| File coverage_metrics_sample_interval_statistics = "${sample}_coverage_metrics.sample_interval_statistics" | |||
| File coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File coverage_metrics_sample_cumulative_coverage_counts = "${sample}_coverage_metrics.sample_cumulative_coverage_counts" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/Realigner.wdl
View File
| @@ -0,0 +1,41 @@ | |||
| task Realigner { | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String db_mills | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo Realigner -k ${dbmills_dir}/${db_mills} ${sample}.sorted.deduped.realigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File realigner_bam = "${sample}.sorted.deduped.realigned.bam" | |||
| File realigner_bam_index = "${sample}.sorted.deduped.realigned.bam.bai" | |||
| } | |||
| } | |||
+ 72
- 0
tasks/benchmark.wdl
View File
| @@ -0,0 +1,72 @@ | |||
| task benchmark { | |||
| File vcf | |||
| File benchmarking_dir | |||
| File ref_dir | |||
| File filtered_bed | |||
| String sample = basename(vcf,".splited.vcf") | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${ref_dir} /cromwell_root/tmp/ | |||
| cp -r ${benchmarking_dir} /cromwell_root/tmp/ | |||
| export HGREF=/cromwell_root/tmp/reference_data/GRCh38.d1.vd1.fa | |||
| cat ${vcf} | grep '#' > header | |||
| cat ${vcf} | grep -v '#' | grep -v '0/0' | grep -v '\./\.'| awk ' | |||
| BEGIN { OFS = "\t" } | |||
| { | |||
| for ( i=9; i<=NF; i++ ) { | |||
| split($i,a,":") ;$i = a[1]; | |||
| } | |||
| } | |||
| { print } | |||
| ' > body | |||
| cat header body > filtered.vcf | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip filtered.vcf -c > ${sample}.rtg.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.rtg.vcf.gz | |||
| if [[ ${sample} =~ "LCL5" ]];then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL5.high.confidence.calls.vcf ${sample}.rtg.vcf.gz -f ${filtered_bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| elif [[ ${sample} =~ "LCL6" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL6.high.confidence.calls.vcf ${sample}.rtg.vcf.gz -f ${filtered_bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| elif [[ ${sample} =~ "LCL7" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL7.high.confidence.calls.vcf ${sample}.rtg.vcf.gz -f ${filtered_bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| elif [[ ${sample} =~ "LCL8" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL8.high.confidence.calls.vcf ${sample}.rtg.vcf.gz -f ${filtered_bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| else | |||
| echo "only for quartet samples" | |||
| fi | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File rtg_vcf = "${sample}.rtg.vcf.gz" | |||
| File rtg_vcf_index = "${sample}.rtg.vcf.gz.tbi" | |||
| File gzip_vcf = "${sample}.vcf.gz" | |||
| File gzip_vcf_index = "${sample}.vcf.gz.tbi" | |||
| File roc_all_csv = "${sample}.roc.all.csv.gz" | |||
| File roc_indel = "${sample}.roc.Locations.INDEL.csv.gz" | |||
| File roc_indel_pass = "${sample}.roc.Locations.INDEL.PASS.csv.gz" | |||
| File roc_snp = "${sample}.roc.Locations.SNP.csv.gz" | |||
| File roc_snp_pass = "${sample}.roc.Locations.SNP.PASS.csv.gz" | |||
| File summary = "${sample}.summary.csv" | |||
| File extended = "${sample}.extended.csv" | |||
| File metrics = "${sample}.metrics.json.gz" | |||
| } | |||
| } | |||
+ 44
- 0
tasks/corealigner.wdl
View File
| @@ -0,0 +1,44 @@ | |||
| task corealigner { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String sample | |||
| String SENTIEON_INSTALL_DIR | |||
| String docker | |||
| String cluster_config | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File tumor_recaled_bam | |||
| File tumor_recaled_bam_index | |||
| File normal_recaled_bam | |||
| File normal_recaled_bam_index | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${tumor_recaled_bam} -i ${normal_recaled_bam} --algo Realigner -k ${db_mills} -k ${dbsnp} ${sample}_corealigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File corealigner_bam = "${sample}_corealigned.bam" | |||
| File corealigner_bam_index = "${sample}_corealigned.bam.bai" | |||
| } | |||
| } | |||
+ 45
- 0
tasks/deduped_Metrics.wdl
View File
| @@ -0,0 +1,45 @@ | |||
| task deduped_Metrics { | |||
| File ref_dir | |||
| File bed | |||
| String SENTIEON_INSTALL_DIR | |||
| String sample | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --interval ${bed} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt --algo QualityYield ${sample}_deduped_QualityYield.txt --algo WgsMetricsAlgo ${sample}_deduped_WgsMetricsAlgo.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File deduped_coverage_metrics_sample_summary = "${sample}_deduped_coverage_metrics.sample_summary" | |||
| File deduped_coverage_metrics_sample_statistics = "${sample}_deduped_coverage_metrics.sample_statistics" | |||
| File deduped_coverage_metrics_sample_interval_statistics = "${sample}_deduped_coverage_metrics.sample_interval_statistics" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_counts = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_counts" | |||
| File deduped_mean_quality = "${sample}_deduped_mq_metrics.txt" | |||
| File deduped_qd_metrics = "${sample}_deduped_qd_metrics.txt" | |||
| File deduped_gc_summary = "${sample}_deduped_gc_summary.txt" | |||
| File deduped_gc_metrics = "${sample}_deduped_gc_metrics.txt" | |||
| File dedeuped_aln_metrics = "${sample}_deduped_aln_metrics.txt" | |||
| File deduped_is_metrics = "${sample}_deduped_is_metrics.txt" | |||
| File deduped_QualityYield = "${sample}_deduped_QualityYield.txt" | |||
| File deduped_wgsmetrics = "${sample}_deduped_WgsMetricsAlgo.txt" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/extract_tables.wdl
View File
| @@ -0,0 +1,41 @@ | |||
| task extract_tables { | |||
| File quality_yield_summary | |||
| File wgs_metrics_summary | |||
| File aln_metrics_summary | |||
| File is_metrics_summary | |||
| File hap | |||
| File fastqc | |||
| File fastqscreen | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/extract_tables.py -quality ${quality_yield_summary} -depth ${wgs_metrics_summary} -aln ${aln_metrics_summary} -is ${is_metrics_summary} -fastqc ${fastqc} -fastqscreen ${fastqscreen} -hap ${hap} -project ${project} | |||
| cat variants.calling.qc.txt | cut -f12- | grep -v 'SNV' | awk '{for(i=1;i<=NF;i++) {sum[i] += $i; sumsq[i] += ($i)^2}} | |||
| END {for (i=1;i<=NF;i++) { | |||
| printf "%f %f \n", sum[i]/NR, sqrt((sumsq[i]-sum[i]^2/NR)/NR)} | |||
| }' >> reference_datasets_aver-std.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File pre_alignment = "pre_alignment.txt" | |||
| File post_alignment = "post_alignment.txt" | |||
| File variant_calling = "variants.calling.qc.txt" | |||
| File precision_recall = "reference_datasets_aver-std.txt" | |||
| } | |||
| } | |||
+ 28
- 0
tasks/fastqc.wdl
View File
| @@ -0,0 +1,28 @@ | |||
| task fastqc { | |||
| File read1 | |||
| File read2 | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| fastqc -t $nt -o ./ ${read1} | |||
| fastqc -t $nt -o ./ ${read2} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File read1_html = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||
| File read1_zip = sub(basename(read1), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||
| File read2_html = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.html") | |||
| File read2_zip = sub(basename(read2), "\\.(fastq|fq)\\.gz$", "_fastqc.zip") | |||
| } | |||
| } | |||
+ 36
- 0
tasks/fastqscreen.wdl
View File
| @@ -0,0 +1,36 @@ | |||
| task fastq_screen { | |||
| File read1 | |||
| File read2 | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| String read1name = basename(read1,".fastq.gz") | |||
| String read2name = basename(read2,".fastq.gz") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${screen_ref_dir} /cromwell_root/tmp/ | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read1} | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --top 100000 --threads $nt ${read2} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File png1 = "${read1name}_screen.png" | |||
| File txt1 = "${read1name}_screen.txt" | |||
| File html1 = "${read1name}_screen.html" | |||
| File png2 = "${read2name}_screen.png" | |||
| File txt2 = "${read2name}_screen.txt" | |||
| File html2 = "${read2name}_screen.html" | |||
| } | |||
| } | |||
+ 35
- 0
tasks/filter_bed.wdl
View File
| @@ -0,0 +1,35 @@ | |||
| task filter_bed { | |||
| File gvcf | |||
| File bed | |||
| File benchmark_region | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${gvcf} | grep '#' > header | |||
| cat ${gvcf} | grep -v '#' > body | |||
| cat body | grep -w '^chr1\|^chr2\|^chr3\|^chr4\|^chr5\|^chr6\|^chr7\|^chr8\|^chr9\|^chr10\|^chr11\|^chr12\|^chr13\|^chr14\|^chr15\|^chr16\|^chr17\|^chr18\|^chr19\|^chr20\|^chr21\|^chr22\|^chrX' > body.filtered | |||
| cat header body.filtered > ${project}.filtered.g.vcf | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${project}.filtered.g.vcf -b ${bed} > body.bed.filtered | |||
| cat header body.bed.filtered > ${project}.chrom.bed.filtered.g.vcf | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${benchmark_region} -b ${bed} > benchmark_region_query_bed.bed | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File filtered_gvcf = "${project}.chrom.bed.filtered.g.vcf" | |||
| File filtered_bed = "benchmark_region_query_bed.bed" | |||
| } | |||
| } | |||
+ 35
- 0
tasks/mapping.wdl
View File
| @@ -0,0 +1,35 @@ | |||
| task mapping { | |||
| File ref_dir | |||
| String fasta | |||
| File fastq_1 | |||
| File fastq_2 | |||
| String SENTIEON_INSTALL_DIR | |||
| String SENTIEON_LICENSE | |||
| String group | |||
| String sample | |||
| String pl | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=${SENTIEON_LICENSE} | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i - | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File sorted_bam = "${sample}.sorted.bam" | |||
| File sorted_bam_index = "${sample}.sorted.bam.bai" | |||
| } | |||
| } | |||
+ 46
- 0
tasks/mendelian.wdl
View File
| @@ -0,0 +1,46 @@ | |||
| task mendelian { | |||
| File family_vcf | |||
| File ref_dir | |||
| String family_name = basename(family_vcf,".family.vcf") | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||
| nt=$(nproc) | |||
| echo -e "${family_name}\tLCL8\t0\t0\t2\t-9\n${family_name}\tLCL7\t0\t0\t1\t-9\n${family_name}\tLCL5\tLCL7\tLCL8\t2\t-9" > ${family_name}.D5.ped | |||
| mkdir VBT_D5 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D5.ped -outDir VBT_D5 -out-prefix ${family_name}.D5 --output-violation-regions -thread-count $nt | |||
| cat VBT_D5/${family_name}.D5_trio.vcf > ${family_name}.D5.vcf | |||
| echo -e "${family_name}\tLCL8\t0\t0\t2\t-9\n${family_name}\tLCL7\t0\t0\t1\t-9\n${family_name}\tLCL6\tLCL7\tLCL8\t2\t-9" > ${family_name}.D6.ped | |||
| mkdir VBT_D6 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D6.ped -outDir VBT_D6 -out-prefix ${family_name}.D6 --output-violation-regions -thread-count $nt | |||
| cat VBT_D6/${family_name}.D6_trio.vcf > ${family_name}.D6.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File D5_ped = "${family_name}.D5.ped" | |||
| File D6_ped = "${family_name}.D6.ped" | |||
| Array[File] D5_mendelian = glob("VBT_D5/*") | |||
| Array[File] D6_mendelian = glob("VBT_D6/*") | |||
| File D5_trio_vcf = "${family_name}.D5.vcf" | |||
| File D6_trio_vcf = "${family_name}.D6.vcf" | |||
| } | |||
| } | |||
+ 26
- 0
tasks/mergeNum.wdl
View File
| @@ -0,0 +1,26 @@ | |||
| task mergeNum { | |||
| Array[File] vcfnumber | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| for i in ${sep=" " vcfnumber} | |||
| do | |||
| cat $i | cut -d':' -f2 | tr '\n' '\t' | sed s'/\t$/\n/g' >> vcfstats | |||
| done | |||
| sed '1i\File\tFailed Filters\tPassed Filters\tSNPs\tMNPs\tInsertions\tDeletions\tIndels\tSame as reference\tSNP Transitions/Transversions\tTotal Het/Hom ratio\tSNP Het/Hom ratio\tMNP Het/Hom ratio\tInsertion Het/Hom ratio\tDeletion Het/Hom ratio\tIndel Het/Hom ratio\tInsertion/Deletion ratio\tIndel/SNP+MNP ratio' vcfstats > vcfstats.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcfstat="vcfstats.txt" | |||
| } | |||
| } | |||
+ 56
- 0
tasks/merge_family.wdl
View File
| @@ -0,0 +1,56 @@ | |||
| task merge_family { | |||
| Array[File] splited_vcf | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| mkdir -p /cromwell_root/tmp/vcf | |||
| cp ${sep=" " splited_vcf} /cromwell_root/tmp/vcf | |||
| for a in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| for b in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| for c in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| for d in /cromwell_root/tmp/vcf/*vcf | |||
| do | |||
| sample_a=$(echo $a | cut -f7 -d_) | |||
| sample_b=$(echo $b | cut -f7 -d_) | |||
| sample_c=$(echo $c | cut -f7 -d_) | |||
| sample_d=$(echo $d | cut -f7 -d_) | |||
| rep_a=$(echo $a | cut -f8 -d_) | |||
| rep_b=$(echo $b | cut -f8 -d_) | |||
| rep_c=$(echo $c | cut -f8 -d_) | |||
| rep_d=$(echo $d | cut -f8 -d_) | |||
| if [ $sample_a == "LCL5" ] && [ $sample_b == "LCL6" ] && [ $sample_c == "LCL7" ] && [ $sample_d == "LCL8" ] && [ $rep_a == $rep_b ] && [ $rep_c == $rep_d ] && [ $rep_b == $rep_c ];then | |||
| cat $a | grep -v '#' > LCL5.body | |||
| cat $b | grep -v '#' | cut -f 10 > LCL6.body | |||
| cat $c | grep -v '#' | cut -f 10 > LCL7.body | |||
| cat $d | grep -v '#' | cut -f 10 > LCL8.body | |||
| echo -e "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5\tLCL6\tLCL7\tLCL8" > header_name | |||
| cat $a | grep '##' | cat - header_name > header | |||
| paste LCL5.body LCL6.body LCL7.body LCL8.body > family.body | |||
| cat header family.body > ${project}_$rep_a.family.vcf | |||
| fi | |||
| done | |||
| done | |||
| done | |||
| done | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| Array[File] family_vcf = glob("*.family.vcf") | |||
| } | |||
| } | |||
+ 35
- 0
tasks/merge_mendelian.wdl
View File
| @@ -0,0 +1,35 @@ | |||
| task merge_mendelian { | |||
| File D5_trio_vcf | |||
| File D6_trio_vcf | |||
| File family_vcf | |||
| String family_name = basename(family_vcf,".family.vcf") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${D5_trio_vcf} | grep -v '##' > ${family_name}.D5.txt | |||
| cat ${D6_trio_vcf} | grep -v '##' > ${family_name}.D6.txt | |||
| cat ${family_vcf} | grep -v '##' | awk ' | |||
| BEGIN { OFS = "\t" } | |||
| NF > 2 && FNR > 1 { | |||
| for ( i=9; i<=NF; i++ ) { | |||
| split($i,a,":") ;$i = a[1]; | |||
| } | |||
| } | |||
| { print } | |||
| ' > ${family_name}.consensus.txt | |||
| python /opt/merge_two_family_with_genotype.py -LCL5 ${family_name}.D5.txt -LCL6 ${family_name}.D6.txt -genotype ${family_name}.consensus.txt -family ${family_name} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File project_mendelian = "${family_name}.txt" | |||
| File project_mendelian_summary = "${family_name}.summary.txt" | |||
| } | |||
| } | |||
+ 47
- 0
tasks/merge_sentieon_metrics.wdl
View File
| @@ -0,0 +1,47 @@ | |||
| task merge_sentieon_metrics { | |||
| Array[File] quality_yield_header | |||
| Array[File] wgs_metrics_algo_header | |||
| Array[File] aln_metrics_header | |||
| Array[File] is_metrics_header | |||
| Array[File] quality_yield_data | |||
| Array[File] wgs_metrics_algo_data | |||
| Array[File] aln_metrics_data | |||
| Array[File] is_metrics_data | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| echo '''Sample''' > sample_column | |||
| cat ${sep=" " quality_yield_header} | sed -n '1,1p' | cat - ${sep=" " quality_yield_data} > quality_yield_all | |||
| ls ${sep=" " quality_yield_data} | cut -d '.' -f1 | cat sample_column - | paste - quality_yield_all > ${project}.quality_yield.txt | |||
| cat ${sep=" " wgs_metrics_algo_header} | sed -n '1,1p' | cat - ${sep=" " wgs_metrics_algo_data} > wgs_metrics_all | |||
| ls ${sep=" " wgs_metrics_algo_data} | cut -d '.' -f1 | cat sample_column - | paste - wgs_metrics_all > ${project}.wgs_metrics_data.txt | |||
| cat ${sep=" " aln_metrics_header} | sed -n '1,1p' | cat - ${sep=" " aln_metrics_data} > aln_metrics_all | |||
| ls ${sep=" " aln_metrics_data} | cut -d '.' -f1 | cat sample_column - | paste - aln_metrics_all > ${project}.aln_metrics.txt | |||
| cat ${sep=" " is_metrics_header} | sed -n '1,1p' | cat - ${sep=" " is_metrics_data} > is_metrics_all | |||
| ls ${sep=" " is_metrics_data} | cut -d '.' -f1 | cat sample_column - | paste - is_metrics_all > ${project}.is_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File quality_yield_summary = "${project}.quality_yield.txt" | |||
| File wgs_metrics_summary = "${project}.wgs_metrics_data.txt" | |||
| File aln_metrics_summary = "${project}.aln_metrics.txt" | |||
| File is_metrics_summary = "${project}.is_metrics.txt" | |||
| } | |||
| } | |||
+ 50
- 0
tasks/multiqc.wdl
View File
| @@ -0,0 +1,50 @@ | |||
| task multiqc { | |||
| Array[File] read1_zip | |||
| Array[File] read2_zip | |||
| Array[File] txt1 | |||
| Array[File] txt2 | |||
| Array[File] summary | |||
| Array[File] zip | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| mkdir -p /cromwell_root/tmp/fastqc | |||
| mkdir -p /cromwell_root/tmp/fastqscreen | |||
| mkdir -p /cromwell_root/tmp/benchmark | |||
| cp ${sep=" " read1_zip} ${sep=" " read2_zip} /cromwell_root/tmp/fastqc | |||
| cp ${sep=" " txt1} ${sep=" " txt2} /cromwell_root/tmp/fastqscreen | |||
| cp ${sep=" " summary} /cromwell_root/tmp/benchmark | |||
| multiqc /cromwell_root/tmp/ | |||
| cat multiqc_data/multiqc_general_stats.txt > multiqc_general_stats.txt | |||
| cat multiqc_data/multiqc_fastq_screen.txt > multiqc_fastq_screen.txt | |||
| cat multiqc_data/multiqc_happy_data.json > multiqc_happy_data.json | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File multiqc_html = "multiqc_report.html" | |||
| Array[File] multiqc_txt = glob("multiqc_data/*") | |||
| File fastqc = "multiqc_general_stats.txt" | |||
| File fastqscreen = "multiqc_fastq_screen.txt" | |||
| File hap = "multiqc_happy_data.json" | |||
| } | |||
| } | |||
+ 29
- 0
tasks/qualimap.wdl
View File
| @@ -0,0 +1,29 @@ | |||
| task qualimap { | |||
| File bam | |||
| File bai | |||
| File bed | |||
| String bamname = basename(bam,".bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| awk 'BEGIN{OFS="\t"}{sub("\r","",$3);print $1,$2,$3,"",0,"."}' ${bed} > new.bed | |||
| /opt/qualimap/qualimap bamqc -bam ${bam} -gff new.bed -outformat PDF:HTML -nt $nt -outdir ${bamname} --java-mem-size=60G | |||
| tar -zcvf ${bamname}_qualimap.zip ${bamname} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File zip = "${bamname}_qualimap.zip" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/quartet_mendelian.wdl
View File
| @@ -0,0 +1,37 @@ | |||
| task quartet_mendelian { | |||
| Array[File] project_mendelian_summary | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| for i in ${sep=" " project_mendelian_summary} | |||
| do | |||
| cat $i | sed -n '2,3p' >> mendelian.summary | |||
| done | |||
| sed '1iFamily\tTotal_Variants\tMendelian_Concordant_Variants\tMendelian_Concordance_Rate' mendelian.summary > mendelian.txt | |||
| cat mendelian.txt | grep 'INDEL' | cut -f4 | grep -v 'Mendelian_Concordance_Rate' | awk '{for(i=1;i<=NF;i++) {sum[i] += $i; sumsq[i] += ($i)^2}} | |||
| END {for (i=1;i<=NF;i++) { | |||
| printf "%f %f \n", sum[i]/NR, sqrt((sumsq[i]-sum[i]^2/NR)/NR)} | |||
| }' >> quartet_indel_aver-std.txt | |||
| cat mendelian.txt | grep 'SNV' | cut -f4 | grep -v 'Mendelian_Concordance_Rate' | awk '{for(i=1;i<=NF;i++) {sum[i] += $i; sumsq[i] += ($i)^2}} | |||
| END {for (i=1;i<=NF;i++) { | |||
| printf "%f %f \n", sum[i]/NR, sqrt((sumsq[i]-sum[i]^2/NR)/NR)} | |||
| }' >> quartet_snv_aver-std.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File mendelian_summary = "mendelian.txt" | |||
| File snv_aver_std = "quartet_snv_aver-std.txt" | |||
| File indel_aver_std = "quartet_indel_aver-std.txt" | |||
| } | |||
| } | |||
+ 47
- 0
tasks/sentieon.wdl
View File
| @@ -0,0 +1,47 @@ | |||
| task sentieon { | |||
| File quality_yield | |||
| File wgs_metrics_algo | |||
| File aln_metrics | |||
| File is_metrics | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| cat ${quality_yield} | sed -n '2,2p' > quality_yield.header | |||
| cat ${quality_yield} | sed -n '3,3p' > ${sample}.quality_yield | |||
| cat ${wgs_metrics_algo} | sed -n '2,2p' > wgs_metrics_algo.header | |||
| cat ${wgs_metrics_algo} | sed -n '3,3p' > ${sample}.wgs_metrics_algo | |||
| cat ${aln_metrics} | sed -n '2,2p' > aln_metrics.header | |||
| cat ${aln_metrics} | sed -n '5,5p' > ${sample}.aln_metrics | |||
| cat ${is_metrics} | sed -n '2,2p' > is_metrics.header | |||
| cat ${is_metrics} | sed -n '3,3p' > ${sample}.is_metrics | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File quality_yield_header = "quality_yield.header" | |||
| File quality_yield_data = "${sample}.quality_yield" | |||
| File wgs_metrics_algo_header = "wgs_metrics_algo.header" | |||
| File wgs_metrics_algo_data = "${sample}.wgs_metrics_algo" | |||
| File aln_metrics_header = "aln_metrics.header" | |||
| File aln_metrics_data = "${sample}.aln_metrics" | |||
| File is_metrics_header = "is_metrics.header" | |||
| File is_metrics_data = "${sample}.is_metrics" | |||
| } | |||
| } | |||
+ 45
- 0
tasks/split_gvcf_files.wdl
View File
| @@ -0,0 +1,45 @@ | |||
| task split_gvcf_files { | |||
| File filtered_gvcf | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${filtered_gvcf} | grep '#CHROM' | sed s'/\t/\n/g' > name | |||
| ncol=`cat name | wc -l` | |||
| sed -i '1,9d' name | |||
| for i in $(seq 10 $ncol); do cat ${filtered_gvcf} | cut -f1-9,$i > $i.splited.vcf; done | |||
| ls *splited.vcf | sort -n | paste - name > rename | |||
| cat rename | while read a b | |||
| do | |||
| mv $a $b.splited.vcf | |||
| sample=$(echo $b | cut -f6 -d_) | |||
| rep=$(echo $b | cut -f7 -d_) | |||
| echo $sample >> quartet_sample | |||
| echo $rep >> quartet_rep | |||
| done | |||
| python /opt/how_many_samples.py -sample quartet_sample -rep quartet_rep | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| Array[File] splited_vcf = glob("*.splited.vcf") | |||
| File sister_tag = "sister_tag" | |||
| File quartet_tag = "quartet_tag" | |||
| } | |||
| } | |||
+ 308
- 0
workflow.wdl
View File
| @@ -0,0 +1,308 @@ | |||
| import "./tasks/mapping.wdl" as mapping | |||
| import "./tasks/Dedup.wdl" as Dedup | |||
| import "./tasks/qualimap.wdl" as qualimap | |||
| import "./tasks/deduped_Metrics.wdl" as deduped_Metrics | |||
| import "./tasks/sentieon.wdl" as sentieon | |||
| import "./tasks/Realigner.wdl" as Realigner | |||
| import "./tasks/BQSR.wdl" as BQSR | |||
| import "./tasks/Haplotyper_gVCF.wdl" as Haplotyper_gVCF | |||
| import "./tasks/GVCFtyper.wdl" as GVCFtyper | |||
| import "./tasks/filter_bed.wdl" as filter_bed | |||
| import "./tasks/split_gvcf_files.wdl" as split_gvcf_files | |||
| import "./tasks/benchmark.wdl" as benchmark | |||
| import "./tasks/multiqc.wdl" as multiqc | |||
| import "./tasks/merge_sentieon_metrics.wdl" as merge_sentieon_metrics | |||
| import "./tasks/extract_tables.wdl" as extract_tables | |||
| import "./tasks/mendelian.wdl" as mendelian | |||
| import "./tasks/merge_mendelian.wdl" as merge_mendelian | |||
| import "./tasks/quartet_mendelian.wdl" as quartet_mendelian | |||
| import "./tasks/fastqc.wdl" as fastqc | |||
| import "./tasks/fastqscreen.wdl" as fastqscreen | |||
| import "./tasks/D5_D6.wdl" as D5_D6 | |||
| import "./tasks/merge_family.wdl" as merge_family | |||
| workflow {{ project_name }} { | |||
| File inputSamplesFile | |||
| Array[Array[File]] inputSamples = read_tsv(inputSamplesFile) | |||
| String SENTIEON_INSTALL_DIR | |||
| String SENTIEON_LICENSE | |||
| String SENTIEONdocker | |||
| String FASTQCdocker | |||
| String FASTQSCREENdocker | |||
| String BENCHMARKdocker | |||
| String MENDELIANdocker | |||
| String DIYdocker | |||
| String MULTIQCdocker | |||
| String BEDTOOLSdocker | |||
| String QUALIMAPdocker | |||
| String fasta | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String db_mills | |||
| File dbsnp_dir | |||
| String dbsnp | |||
| File bed | |||
| File benchmark_region | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| File benchmarking_dir | |||
| String project | |||
| String disk_size | |||
| String BIGcluster_config | |||
| String SMALLcluster_config | |||
| scatter (quartet in inputSamples){ | |||
| call mapping.mapping as mapping { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| SENTIEON_LICENSE=SENTIEON_LICENSE, | |||
| group=quartet[2], | |||
| sample=quartet[2], | |||
| pl="ILLUMINAL", | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| fastq_1=quartet[0], | |||
| fastq_2=quartet[1], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen { | |||
| input: | |||
| read1=quartet[0], | |||
| read2=quartet[1], | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf, | |||
| docker=FASTQSCREENdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| call Dedup.Dedup as Dedup { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=mapping.sorted_bam, | |||
| sorted_bam_index=mapping.sorted_bam_index, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call qualimap.qualimap as qualimap { | |||
| input: | |||
| bam=Dedup.Dedup_bam, | |||
| bai=Dedup.Dedup_bam_index, | |||
| bed=bed, | |||
| docker=QUALIMAPdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as deduped_Metrics { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| bed=bed, | |||
| Dedup_bam=Dedup.Dedup_bam, | |||
| Dedup_bam_index=Dedup.Dedup_bam_index, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call sentieon.sentieon as sentieon { | |||
| input: | |||
| quality_yield=deduped_Metrics.deduped_QualityYield, | |||
| wgs_metrics_algo=deduped_Metrics.deduped_wgsmetrics, | |||
| aln_metrics=deduped_Metrics.dedeuped_aln_metrics, | |||
| is_metrics=deduped_Metrics.deduped_is_metrics, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Realigner.Realigner as Realigner { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=Dedup.Dedup_bam, | |||
| Dedup_bam_index=Dedup.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call BQSR.BQSR as BQSR { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| realigned_bam=Realigner.realigner_bam, | |||
| realigned_bam_index=Realigner.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call Haplotyper_gVCF.Haplotyper_gVCF as Haplotyper_gVCF { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=BQSR.recaled_bam, | |||
| recaled_bam_index=BQSR.recaled_bam_index, | |||
| sample=quartet[2], | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| } | |||
| call GVCFtyper.GVCFtyper as GVCFtyper { | |||
| input: | |||
| ref_dir=ref_dir, | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| vcf=Haplotyper_gVCF.vcf, | |||
| vcf_idx=Haplotyper_gVCF.vcf_idx, | |||
| project=project, | |||
| docker=SENTIEONdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call filter_bed.filter_bed as filter_bed { | |||
| input: | |||
| gvcf=GVCFtyper.gvcf, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| docker=BEDTOOLSdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call split_gvcf_files.split_gvcf_files as split_gvcf_files { | |||
| input: | |||
| filtered_gvcf=filter_bed.filtered_gvcf, | |||
| docker=DIYdocker, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| Array[File] single_gvcf = split_gvcf_files.splited_vcf | |||
| scatter (idx in range(length(single_gvcf))) { | |||
| call benchmark.benchmark as benchmark { | |||
| input: | |||
| vcf=single_gvcf[idx], | |||
| benchmarking_dir=benchmarking_dir, | |||
| filtered_bed=filter_bed.filtered_bed, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| call merge_sentieon_metrics.merge_sentieon_metrics as merge_sentieon_metrics { | |||
| input: | |||
| quality_yield_header=sentieon.quality_yield_header, | |||
| wgs_metrics_algo_header=sentieon.wgs_metrics_algo_header, | |||
| aln_metrics_header=sentieon.aln_metrics_header, | |||
| is_metrics_header=sentieon.is_metrics_header, | |||
| quality_yield_data=sentieon.quality_yield_data, | |||
| wgs_metrics_algo_data=sentieon.wgs_metrics_algo_data, | |||
| aln_metrics_data=sentieon.aln_metrics_data, | |||
| is_metrics_data=sentieon.is_metrics_data, | |||
| project=project, | |||
| docker=MULTIQCdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| Boolean sister_tag = read_boolean(split_gvcf_files.sister_tag) | |||
| Boolean quartet_tag = read_boolean(split_gvcf_files.quartet_tag) | |||
| if (sister_tag) { | |||
| call D5_D6.D5_D6 as D5_D6 { | |||
| input: | |||
| splited_vcf=split_gvcf_files.splited_vcf, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size, | |||
| } | |||
| } | |||
| if (quartet_tag) { | |||
| call merge_family.merge_family as merge_family { | |||
| input: | |||
| splited_vcf=split_gvcf_files.splited_vcf, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size, | |||
| } | |||
| Array[File] family_vcfs = merge_family.family_vcf | |||
| scatter (idx in range(length(family_vcfs))) { | |||
| call mendelian.mendelian as mendelian { | |||
| input: | |||
| family_vcf=family_vcfs[idx], | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=MENDELIANdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call merge_mendelian.merge_mendelian as merge_mendelian { | |||
| input: | |||
| D5_trio_vcf=mendelian.D5_trio_vcf, | |||
| D6_trio_vcf=mendelian.D6_trio_vcf, | |||
| family_vcf=family_vcfs[idx], | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| call quartet_mendelian.quartet_mendelian as quartet_mendelian { | |||
| input: | |||
| project_mendelian_summary=merge_mendelian.project_mendelian_summary, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| } | |||
Loading…