
 LUYAO REN
5 years ago
LUYAO REN
5 years ago
27 changed files with 1088 additions and 109 deletions
+ 33
- 0
LCL5.tsv
View File
| Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ2000_WGE_LCL5_1_20190402 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ2000_WGE_LCL5_1_20190402_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ2000_WGE_LCL5_2_20190402 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ2000_WGE_LCL5_2_20190402_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ500_BGI_LCL5_1_20180328 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ500_BGI_LCL5_1_20180328_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ500_BGI_LCL5_2_20180328 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ500_BGI_LCL5_2_20180328_hc.vcf | |||||
| Quartet_DNA_BGI_SEQ500_BGI_LCL5_3_20180328 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_BGI_SEQ500_BGI_LCL5_3_20180328_hc.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_BRG_LCL5_1_20171024 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_BRG_LCL5_1_20171024_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_GAC_LCL5_1_20171025 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_GAC_LCL5_1_20171025_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_NVG_LCL5_1_20171024 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_NVG_LCL5_1_20171024_RAW.vcf | |||||
| Quartet_DNA_ILM_Nova_WUX_LCL5_1_20171024 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_Nova_WUX_LCL5_1_20171024_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403_hc.vcf | |||||
| Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403_hc.vcf | |||||
| Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403_hc.vcf | |||||
| Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_4_20180703 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_4_20180703_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_5_20180703 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_5_20180703_RAW.vcf | |||||
| Quartet_DNA_ILM_XTen_WUX_LCL5_6_20180703 oss://chinese-quartet/quartet-result-data/FDU/VCF/Quartet_DNA_ILM_XTen_WUX_LCL5_6_20180703_RAW.vcf |
+ 25
- 109
README.md
View File
| #高置信位点的整合 | |||||
| #高置信位点整合Part2——从VCF和BAM文件中提取突变位点的信息 | |||||
| ## 一、流程 | |||||
| > Author: Run Luyao | |||||
| > | |||||
| > E-mail:18110700050@fudan.edu.cn | |||||
| > | |||||
| > Git: http://choppy.3steps.cn/renluyao/HCC_Extract_Info.git | |||||
| > | |||||
| > Last Updates: 11/9/2019 | |||||
| ## 二、模块 | |||||
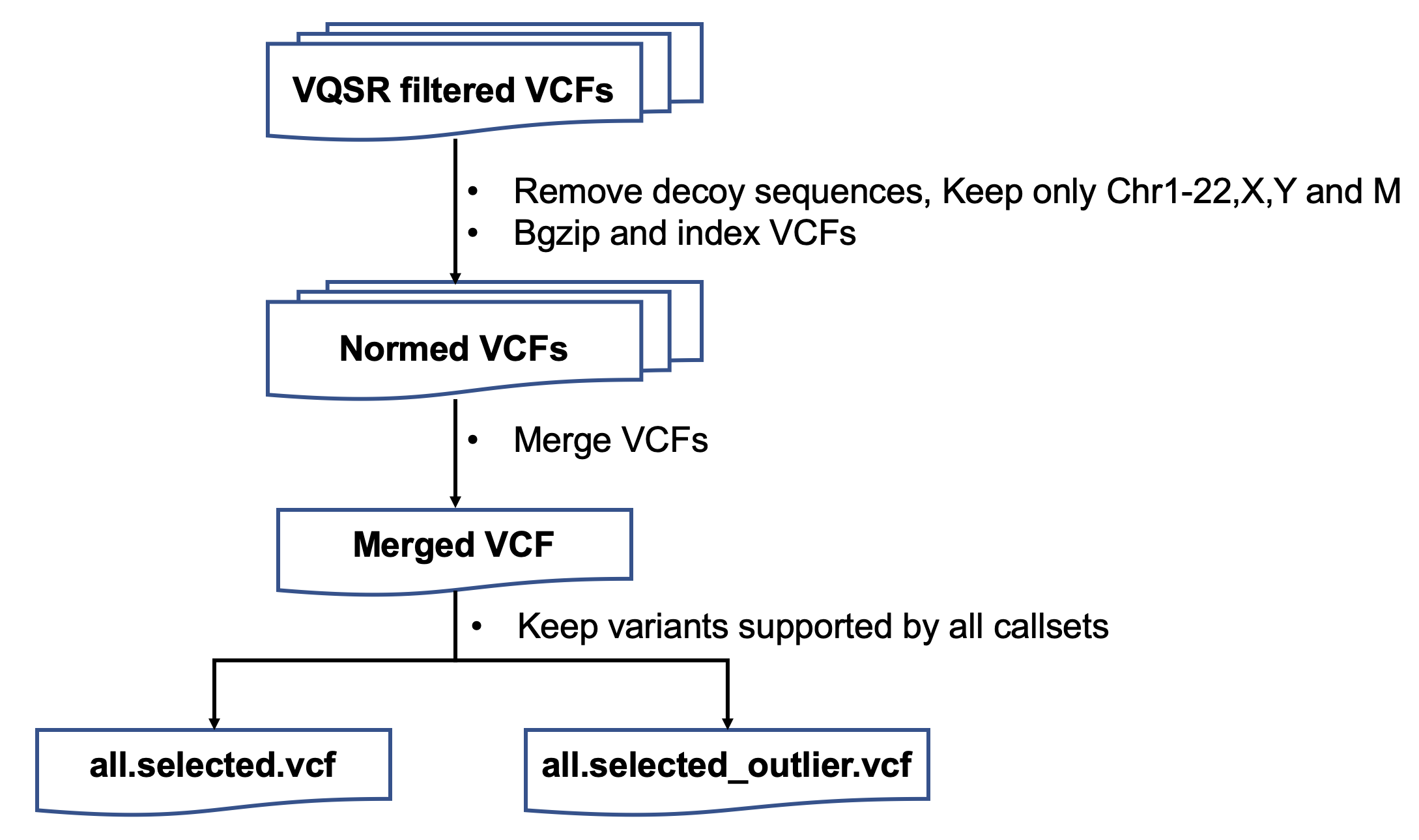
| ###1. 选择被所有callset支持的SNVs和Indels | |||||
| http://choppy.3steps.cn/renluyao/HCC_Select_HF_mutations.git | |||||
| ### 2.从VCF文件和bam文件中提取SNVs和Indel信息 | |||||
| ## 4. Extract SNPs and Indels information | |||||
| (1) Separate the SNVs and Indels | |||||
| ## 安装指南 | |||||
| ```bash | ```bash | ||||
| vcftools --gzvcf sample.filtered.normed.vcf.gz --remove-indels --recode --recode-INFO-all --out sample.filtered.normed.snv | |||||
| vcftools --gzvcf sample.filtered.normed.vcf.gz --keep-only-indels --recode --recode-INFO-all --out sample.filtered.normed.indel | |||||
| rtg bgzip sample.filtered.normed.snv.vcf | |||||
| rtg index sample.filtered.normed.snv.vcf.gz | |||||
| rtg bgzip sample.filtered.normed.indel.vcf | |||||
| rtg index sample.filtered.normed.indel.vcf.gz | |||||
| # 激活choppy环境 | |||||
| source activate choppy | |||||
| # 安装app | |||||
| choppy install renluyao/HCC_Extract_Info | |||||
| ``` | ``` | ||||
| (2) Separate training and testing vcf files | |||||
| ```bash | |||||
| rtg vcffilter -i sample.filtered.normed.snv.vcf.gz -o sample.filtered.normed.snv.train.vcf.gz --include-vcf=all.filtered.vcf.gz | |||||
| rtg vcffilter -i sample.filtered.normed.snv.vcf.gz -o sample.filtered.normed.snv.test.vcf.gz --exclude-vcf=all.filtered.vcf.gz | |||||
| rtg vcffilter -i sample.filtered.normed.indel.vcf.gz -o sample.filtered.normed.indel.train.vcf.gz --include-vcf=all.filtered.vcf.gz | |||||
| rtg vcffilter -i sample.filtered.normed.indel.vcf.gz -o sample.filtered.normed.indel.test.vcf.gz --exclude-vcf=all.filtered.vcf.gz | |||||
| ``` | |||||
| (3) Extract the information from VCF file | |||||
| ```bash | |||||
| gzip -d sample.filtered.normed.snv.train.vcf.gz | |||||
| gzip -d sample.filtered.normed.snv.test.vcf.gz | |||||
| gzip -d sample.filtered.normed.indel.train.vcf.gz | |||||
| gzip -d sample.filtered.normed.indel.test.vcf.gz | |||||
| python extract_vcf_information.py -i sample.filtered.normed.snv.train.vcf -o sample.filtered.normed.snv.train.txt | |||||
| python extract_vcf_information.py -i sample.filtered.normed.snv.test.vcf -o sample.filtered.normed.snv.test.txt | |||||
| python extract_vcf_information.py -i sample.filtered.normed.indel.train.vcf -o sample.filtered.normed.indel.train.txt | |||||
| python extract_vcf_information.py -i sample.filtered.normed.indel.train.vcf -o sample.filtered.normed.indel.train.txt | |||||
| ``` | |||||
| (4)Extract the information from bam-readcount | |||||
| ```bash | |||||
| ## get bed | |||||
| # snv | |||||
| cat sample.filtered.normed.snv.train.vcf | sed '/^#/d' | awk '{print $1"\t"$2"\t"$2"\t"$4"\t"$5}' > sample.filtered.normed.snv.train.bed | |||||
| cat sample.filtered.normed.snv.test.vcf | sed '/^#/d' | awk '{print $1"\t"$2"\t"$2"\t"$4"\t"$5}' > sample.filtered.normed.snv.test.bed | |||||
| #indel | |||||
| python bed_for_bamReadcount.py -i sample.filtered.normed.indel.train.vcf -o sample.filtered.normed.indel.train | |||||
| python bed_for_bamReadcount.py -i sample.filtered.normed.indel.test.vcf -o sample.filtered.normed.indel.test | |||||
| ## bam-readcount | |||||
| #snv | |||||
| bam-readcount -f reference.fa -l sample.filtered.normed.snv.train.bed sample.bam -b 20> sample.filtered.normed.snv.train.readcount | |||||
| bam-readcount -f reference.fa -l sample.filtered.normed.snv.test.bed sample.bam -b 20 > sample.filtered.normed.snv.test.readcount | |||||
| #indel | |||||
| bam-readcount -f reference.fa -l sample.filtered.normed.indel.train.bed sample.bam -i -b 20 > sample.filtered.normed.indel.train.readcount | |||||
| bam-readcount -f reference.fa -l sample.filtered.normed.indel.test.bed sample.bam -i -b 20 > sample.filtered.normed.indel.test.readcount | |||||
| ``` | |||||
| (5) Parse bam-readcount information and combine with vcf information | |||||
| ```bash | |||||
| # add alt in to bam-readcount | |||||
| awk 'NR==FNR{a[$1,$2]=$5;next} ($1,$2) in a{print $0, a[$1,$2]}' OFS="\t" sample.filtered.normed.indel.train.bed sample.filtered.normed.indel.train.readcount > sample.filtered.normed.indel.train.bamReadcount | |||||
| awk 'NR==FNR{a[$1,$2]=$5;next} ($1,$2) in a{print $0, a[$1,$2]}' OFS="\t" sample.filtered.normed.indel.test.bed sample.filtered.normed.indel.test.readcount > sample.filtered.normed.indel.test.bamReadcount | |||||
| awk 'NR==FNR{a[$1,$2]=$5;next} ($1,$2) in a{print $0, a[$1,$2]}' OFS="\t" sample.filtered.normed.snv.train.bed sample.filtered.normed.snv.train.readcount > sample.filtered.normed.snv.train.bamReadcount | |||||
| awk 'NR==FNR{a[$1,$2]=$5;next} ($1,$2) in a{print $0, a[$1,$2]}' OFS="\t" sample.filtered.normed.snv.test.bed sample.filtered.normed.snv.test.readcount > sample.filtered.normed.snv.test.bamReadcount | |||||
| ``` | |||||
| ## 5. Train machine-learning models on every call sets | |||||
| Novelty detection <https://scikit-learn.org/stable/modules/outlier_detection.html#outlier-detection> | |||||
| Example <https://scikit-learn.org/stable/auto_examples/svm/plot_oneclass.html#sphx-glr-auto-examples-svm-plot-oneclass-py> | |||||
| Setting tips <https://scikit-learn.org/stable/modules/svm.html#svm-outlier-detection> | |||||
| (1) Train SNVs model | |||||
| ## App概述 | |||||
| (2) Train Indels model | |||||
| 当我们拿到测序结果的时候,没有办法判断哪些是真的突变位点,哪些是假阳性 | |||||
| ## 流程与参数 | |||||
| 两个问题: | |||||
| ## 6. Intergrate the information | |||||
| 1. MNP | |||||
| 2. merge之后的pos相同,但是ref和alt不同 | |||||
| add info indicates which platform support the variants, how many sequencing sites, replicates, mapper and callers support the variants | |||||
| 是不是gatk的问题,尝试用bcftools norm | |||||
| <https://github.com/brentp/vcfanno/> | |||||
| ## App输入变量与输入文件 | |||||
| ## App输出文件 | |||||
| ## 结果展示与解读 | |||||
| ## 7. WDL | |||||
| ## CHANGELOG | |||||
| <https://gatkforums.broadinstitute.org/wdl/discussion/6716/scatter-gather-parallelism>q | |||||
| ## FAQ | |||||
BIN
codescripts/.DS_Store
View File
+ 68
- 0
codescripts/bed_for_bamReadcount.py
View File
| import sys,getopt | |||||
| import os | |||||
| import re | |||||
| import fileinput | |||||
| def usage(): | |||||
| print( | |||||
| """ | |||||
| Usage: python bed_for_bamReadcount.py -i input_vcf_file -o prefix | |||||
| This script selects SNPs and Indels supported by all callsets. | |||||
| Please notice that bam-readcount only takes in 1-based coordinates. | |||||
| Input: | |||||
| -i a vcf file | |||||
| Output: | |||||
| -o a indel bed file for bam-readcount | |||||
| """) | |||||
| # select supported small variants | |||||
| def process(oneLine): | |||||
| m = re.match('^\#',oneLine) | |||||
| if m is not None: | |||||
| pass | |||||
| else: | |||||
| line = oneLine.rstrip() | |||||
| strings = line.strip().split('\t') | |||||
| # convert the position to bed file for bam-readcount | |||||
| # deletion | |||||
| if len(strings[3]) > 1 and len(strings[4]) == 1: | |||||
| pos = int(strings[1]) + 1 | |||||
| outline = strings[0] + '\t' + str(pos) + '\t' + str(pos) + '\t' + strings[3] + '\t' + strings[4]+'\n' | |||||
| outINDEL.write(outline) | |||||
| # insertion | |||||
| elif len(strings[3]) == 1 and len(strings[4]) > 1 and (',' not in strings[4]): | |||||
| outline = strings[0] + '\t' + strings[1] + '\t' + strings[1] + '\t' + strings[3] + '\t' + strings[4] + '\n' | |||||
| outINDEL.write(outline) | |||||
| else: | |||||
| outMNP.write(oneLine) | |||||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||||
| for op,value in opts: | |||||
| if op == "-i": | |||||
| inputFile=value | |||||
| elif op == "-o": | |||||
| prefix=value | |||||
| elif op == "-h": | |||||
| usage() | |||||
| sys.exit() | |||||
| if len(sys.argv[1:]) < 3: | |||||
| usage() | |||||
| sys.exit() | |||||
| INDELname = prefix + '.bed' | |||||
| MNPname = prefix + '_MNP.txt' | |||||
| outINDEL = open(INDELname,'w') | |||||
| outMNP = open(MNPname,'w') | |||||
| for line in fileinput.input(inputFile): | |||||
| process(line) | |||||
| outINDEL.close() | |||||
| outMNP.close() | |||||
+ 96
- 0
codescripts/extract_vcf_information.py
View File
| import sys,getopt | |||||
| import os | |||||
| import re | |||||
| import fileinput | |||||
| import pandas as pd | |||||
| def usage(): | |||||
| print( | |||||
| """ | |||||
| Usage: python extract_vcf_information.py -i input_merged_vcf_file -o parsed_file | |||||
| This script will extract SNVs and Indels information from the vcf files and output a tab-delimited files. | |||||
| Input: | |||||
| -i the selected vcf file | |||||
| Output: | |||||
| -o tab-delimited parsed file | |||||
| """) | |||||
| # select supported small variants | |||||
| def process(oneLine): | |||||
| line = oneLine.rstrip() | |||||
| strings = line.strip().split('\t') | |||||
| infoParsed = parse_INFO(strings[7]) | |||||
| formatKeys = strings[8].split(':') | |||||
| formatValues = strings[9].split(':') | |||||
| for i in range(0,len(formatKeys) -1) : | |||||
| if formatKeys[i] == 'AD': | |||||
| ra = formatValues[i].split(',') | |||||
| infoParsed['RefDP'] = ra[0] | |||||
| infoParsed['AltDP'] = ra[1] | |||||
| if (int(ra[1]) + int(ra[0])) != 0: | |||||
| infoParsed['af'] = float(int(ra[1])/(int(ra[1]) + int(ra[0]))) | |||||
| else: | |||||
| pass | |||||
| else: | |||||
| infoParsed[formatKeys[i]] = formatValues[i] | |||||
| infoParsed['chromo'] = strings[0] | |||||
| infoParsed['pos'] = strings[1] | |||||
| infoParsed['id'] = strings[2] | |||||
| infoParsed['ref'] = strings[3] | |||||
| infoParsed['alt'] = strings[4] | |||||
| infoParsed['qual'] = strings[5] | |||||
| return infoParsed | |||||
| def parse_INFO(info): | |||||
| strings = info.strip().split(';') | |||||
| keys = [] | |||||
| values = [] | |||||
| for i in strings: | |||||
| kv = i.split('=') | |||||
| if kv[0] == 'DB': | |||||
| keys.append('DB') | |||||
| values.append('1') | |||||
| elif kv[0] == 'AF': | |||||
| pass | |||||
| elif kv[0] == 'POSITIVE_TRAIN_SITE': | |||||
| pass | |||||
| elif kv[0] == 'NEGATIVE_TRAIN_SITE': | |||||
| pass | |||||
| else: | |||||
| keys.append(kv[0]) | |||||
| values.append(kv[1]) | |||||
| infoDict = dict(zip(keys, values)) | |||||
| return infoDict | |||||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||||
| for op,value in opts: | |||||
| if op == "-i": | |||||
| inputFile=value | |||||
| elif op == "-o": | |||||
| outputFile=value | |||||
| elif op == "-h": | |||||
| usage() | |||||
| sys.exit() | |||||
| if len(sys.argv[1:]) < 3: | |||||
| usage() | |||||
| sys.exit() | |||||
| allDict = [] | |||||
| for line in fileinput.input(inputFile): | |||||
| m = re.match('^\#',line) | |||||
| if m is not None: | |||||
| pass | |||||
| else: | |||||
| oneDict = process(line) | |||||
| allDict.append(oneDict) | |||||
| allTable = pd.DataFrame(allDict) | |||||
| allTable.to_csv(outputFile,sep='\t',index=False) | |||||
+ 109
- 0
codescripts/oneClass.py
View File
| # import modules | |||||
| import numpy as np | |||||
| import pandas as pd | |||||
| from sklearn import svm | |||||
| from sklearn import preprocessing | |||||
| import sys, argparse, os | |||||
| from vcf2bed import position_to_bed,padding_region | |||||
| parser = argparse.ArgumentParser(description="this script is to preform one calss svm on each chromosome") | |||||
| parser.add_argument('-train', '--trainDataset', type=str, help='training dataset generated from extracting vcf information part, with mutaitons supported by callsets', required=True) | |||||
| parser.add_argument('-test', '--testDataset', type=str, help='testing dataset generated from extracting vcf information part, with mutaitons not called by all callsets', required=True) | |||||
| parser.add_argument('-name', '--sampleName', type=str, help='sample name for output file name', required=True) | |||||
| args = parser.parse_args() | |||||
| # Rename input: | |||||
| train_input = args.trainDataset | |||||
| test_input = args.testDataset | |||||
| sample_name = args.sampleName | |||||
| # default columns, which will be included in the included in the calssifier | |||||
| chromosome = ['chr1','chr2','chr3','chr4','chr5','chr6','chr7','chr8','chr9','chr10','chr11','chr12','chr13','chr14','chr15' ,'chr16','chr17','chr18','chr19','chr20','chr21','chr22','chrX','chrY'] | |||||
| feature_heter_cols = ['AltDP','BaseQRankSum','DB','DP','FS','GQ','MQ','MQRankSum','QD','ReadPosRankSum','RefDP','SOR','af'] | |||||
| feature_homo_cols = ['AltDP','DB','DP','FS','GQ','MQ','QD','RefDP','SOR','af'] | |||||
| # import datasets sepearate the records with or without BaseQRankSum annotation, etc. | |||||
| def load_dat(dat_file_name): | |||||
| dat = pd.read_table(dat_file_name) | |||||
| dat['DB'] = dat['DB'].fillna(0) | |||||
| dat = dat[dat['DP'] != 0] | |||||
| dat['af'] = dat['AltDP']/(dat['AltDP'] + dat['RefDP']) | |||||
| homo_rows = dat[dat['BaseQRankSum'].isnull()] | |||||
| heter_rows = dat[dat['BaseQRankSum'].notnull()] | |||||
| return homo_rows,heter_rows | |||||
| train_homo,train_heter = load_dat(train_input) | |||||
| test_homo,test_heter = load_dat(test_input) | |||||
| clf = svm.OneClassSVM(nu=0.05,kernel='rbf', gamma='auto_deprecated',cache_size=500) | |||||
| def prepare_dat(train_dat,test_dat,feature_cols,chromo): | |||||
| chr_train = train_dat[train_dat['chromo'] == chromo] | |||||
| chr_test = test_dat[test_dat['chromo'] == chromo] | |||||
| train_dat = chr_train.loc[:,feature_cols] | |||||
| test_dat = chr_test.loc[:,feature_cols] | |||||
| train_dat_scaled = preprocessing.scale(train_dat) | |||||
| test_dat_scaled = preprocessing.scale(test_dat) | |||||
| return chr_test,train_dat_scaled,test_dat_scaled | |||||
| def oneclass(X_train,X_test,chr_test): | |||||
| clf.fit(X_train) | |||||
| y_pred_test = clf.predict(X_test) | |||||
| test_true_dat = chr_test[y_pred_test == 1] | |||||
| test_false_dat = chr_test[y_pred_test == -1] | |||||
| return test_true_dat,test_false_dat | |||||
| predicted_true = pd.DataFrame(columns=train_homo.columns) | |||||
| predicted_false = pd.DataFrame(columns=train_homo.columns) | |||||
| for chromo in chromosome: | |||||
| # homo datasets | |||||
| chr_test_homo,X_train_homo,X_test_homo = prepare_dat(train_homo,test_homo,feature_homo_cols,chromo) | |||||
| test_true_homo,test_false_homo = oneclass(X_train_homo,X_test_homo,chr_test_homo) | |||||
| predicted_true = predicted_true.append(test_true_homo) | |||||
| predicted_false = predicted_false.append(test_false_homo) | |||||
| # heter datasets | |||||
| chr_test_heter,X_train_heter,X_test_heter = prepare_dat(train_heter,test_heter,feature_heter_cols,chromo) | |||||
| test_true_heter,test_false_heter = oneclass(X_train_heter,X_test_heter,chr_test_heter) | |||||
| predicted_true = predicted_true.append(test_true_heter) | |||||
| predicted_false = predicted_false.append(test_false_heter) | |||||
| predicted_true_filename = sample_name + '_predicted_true.txt' | |||||
| predicted_false_filename = sample_name + '_predicted_false.txt' | |||||
| predicted_true.to_csv(predicted_true_filename,sep='\t',index=False) | |||||
| predicted_false.to_csv(predicted_false_filename,sep='\t',index=False) | |||||
| # output the bed file and padding bed region 50bp | |||||
| predicted_true_bed_filename = sample_name + '_predicted_true.bed' | |||||
| predicted_false_bed_filename = sample_name + '_predicted_false.bed' | |||||
| padding_filename = sample_name + '_padding.bed' | |||||
| predicted_true_bed = open(predicted_true_bed_filename,'w') | |||||
| predicted_false_bed = open(predicted_false_bed_filename,'w') | |||||
| padding = open(padding_filename,'w') | |||||
| # | |||||
| for index,row in predicted_false.iterrows(): | |||||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||||
| predicted_false_bed.write(outline_pos) | |||||
| chromo,pad_pos1,pad_pos2,pad_pos3,pad_pos4 = padding_region(chromo,pos1,pos2,50) | |||||
| outline_pad_1 = chromo + '\t' + str(pad_pos1) + '\t' + str(pad_pos2) + '\n' | |||||
| outline_pad_2 = chromo + '\t' + str(pad_pos3) + '\t' + str(pad_pos4) + '\n' | |||||
| padding.write(outline_pad_1) | |||||
| padding.write(outline_pad_2) | |||||
| for index,row in predicted_true.iterrows(): | |||||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||||
| predicted_true_bed.write(outline_pos) | |||||
+ 62
- 0
codescripts/select_small_variants_supported_by_all_callsets.py
View File
| import sys,getopt | |||||
| import os | |||||
| import re | |||||
| import fileinput | |||||
| def usage(): | |||||
| print( | |||||
| """ | |||||
| Usage: python select_small_variants_supported_by_all_callsets.py -i input_merged_vcf_file -o prefix | |||||
| This script selects SNPs and Indels supported by all callsets. | |||||
| Input: | |||||
| -i a merged vcf file | |||||
| Output: | |||||
| -o a vcf file containd the selected SNPs and Indels | |||||
| """) | |||||
| # select supported small variants | |||||
| def process(oneLine): | |||||
| m = re.match('^\#',oneLine) | |||||
| if m is not None: | |||||
| outVCF.write(oneLine) | |||||
| OUTname.write(oneLine) | |||||
| else: | |||||
| line = oneLine.rstrip() | |||||
| strings = line.strip().split('\t') | |||||
| gt = [i.split(':', 1)[0] for i in strings[9:len(strings)]] | |||||
| if all(e == gt[0] for e in gt) and (gt[0] != '.'): | |||||
| # output the record to vcf | |||||
| outVCF.write(oneLine) | |||||
| else: | |||||
| OUTname.write(oneLine) | |||||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||||
| for op,value in opts: | |||||
| if op == "-i": | |||||
| inputFile=value | |||||
| elif op == "-o": | |||||
| prefix=value | |||||
| elif op == "-h": | |||||
| usage() | |||||
| sys.exit() | |||||
| if len(sys.argv[1:]) < 3: | |||||
| usage() | |||||
| sys.exit() | |||||
| VCFname = prefix + '.vcf' | |||||
| OUTname = prefix + '_outlier.vcf' | |||||
| outVCF = open(VCFname,'w') | |||||
| OUTname = open(OUTname,'w') | |||||
| for line in fileinput.input(inputFile): | |||||
| process(line) | |||||
| outVCF.close() | |||||
| OUTname.close() | |||||
+ 36
- 0
codescripts/vcf2bed.py
View File
| import re | |||||
| def position_to_bed(chromo,pos,ref,alt): | |||||
| # snv | |||||
| # Start cooridinate BED = start coordinate VCF - 1 | |||||
| # End cooridinate BED = start coordinate VCF | |||||
| if len(ref) == 1 and len(alt) == 1: | |||||
| StartPos = int(pos) -1 | |||||
| EndPos = int(pos) | |||||
| # deletions | |||||
| # Start cooridinate BED = start coordinate VCF - 1 | |||||
| # End cooridinate BED = start coordinate VCF + (reference length - alternate length) | |||||
| elif len(ref) > 1 and len(alt) == 1: | |||||
| StartPos = int(pos) - 1 | |||||
| EndPos = int(pos) + (len(ref) - 1) | |||||
| #insertions | |||||
| # For insertions: | |||||
| # Start cooridinate BED = start coordinate VCF - 1 | |||||
| # End cooridinate BED = start coordinate VCF + (alternate length - reference length) | |||||
| else: | |||||
| StartPos = int(pos) - 1 | |||||
| EndPos = int(pos) + (len(alt) - 1) | |||||
| return chromo,StartPos,EndPos | |||||
| def padding_region(chromo,pos1,pos2,padding): | |||||
| StartPos1 = pos1 - padding | |||||
| EndPos1 = pos1 | |||||
| StartPos2 = pos2 | |||||
| EndPos2 = pos2 + padding | |||||
| return chromo,StartPos1,EndPos1,StartPos2,EndPos2 |
+ 1
- 0
family.tsv
View File
| #LCL5vcf LCL6vcf LCL7vcf LCL8vcf LCL5name LCL6name LCL7name LCL8name familyname |
+ 14
- 0
inputs
View File
| { | |||||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||||
| "{{ project_name }}.mendelian.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/vbt:v1.1", | |||||
| "{{ project_name }}.disk_size": "100", | |||||
| "{{ project_name }}.merge.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||||
| "{{ project_name }}.zipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||||
| "{{ project_name }}.inputSamplesFile": "{{ inputSamplesFile }}", | |||||
| "{{ project_name }}.LCL6variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||||
| "{{ project_name }}.cluster_config": "OnDemand bcs.a2.large img-ubuntu-vpc", | |||||
| "{{ project_name }}.LCL7variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||||
| "{{ project_name }}.LCL5variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||||
| "{{ project_name }}.LCL8variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||||
| "{{ project_name }}.ref_dir": "oss://chinese-quartet/quartet-storage-data/reference_data/" | |||||
| } |
BIN
pictures/.DS_Store
View File
BIN
pictures/workflow.png
View File
{kind=link}
| Before | After |
|---|---|
|
|
| Width: 2178 | Height: 1290 | Size: 180KB |
BIN
tasks/.DS_Store
View File
+ 34
- 0
tasks/ExtractVCFinfo.wdl
View File
| task ExtractVCFinfo { | |||||
| File snv_train | |||||
| File snv_test | |||||
| File indel_train | |||||
| File indel_test | |||||
| String snv_train_sampleName = basename(snv_train,".vcf") | |||||
| String snv_test_sampleName = basename(snv_test,".vcf") | |||||
| String indel_train_sampleName = basename(indel_train,".vcf") | |||||
| String indel_test_sampleName = basename(indel_test,".vcf") | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| python /opt/extract_vcf_information.py -i ${snv_train} -o ${snv_train_sampleName}.txt | |||||
| python /opt/extract_vcf_information.py -i ${snv_test} -o ${snv_test_sampleName}.txt | |||||
| python /opt/extract_vcf_information.py -i ${indel_train} -o ${indel_train_sampleName}.txt | |||||
| python /opt/extract_vcf_information.py -i ${indel_test} -o ${indel_test_sampleName}.txt | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File snv_train_vcf = "${snv_train_sampleName}.txt" | |||||
| File snv_test_vcf = "${snv_test_sampleName}.txt" | |||||
| File indel_train_vcf = "${indel_train_sampleName}.txt" | |||||
| File indel_test_vcf = "${indel_test_sampleName}.txt" | |||||
| } | |||||
| } |
+ 27
- 0
tasks/KeepVar.wdl
View File
| task KeepVar { | |||||
| File violation_merged_vcf | |||||
| File consistent_merged_vcf | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| python /opt/select_small_variants_supported_by_all_callsets.py -i ${violation_merged_vcf} -o violation.all.selected | |||||
| python /opt/select_small_variants_supported_by_all_callsets.py -i ${consistent_merged_vcf} -o consistent.all.selected | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File violation_keeped_vcf = "violation.all.selected.vcf" | |||||
| File violation_outlier_vcf = "violation.all.selected_outlier.vcf" | |||||
| File consistent_keeped_vcf = "consistent.all.selected.vcf" | |||||
| File consistent_outlier_vcf = "consistent.all.selected_outlier.vcf" | |||||
| } | |||||
| } | |||||
+ 69
- 0
tasks/SepSnvIndel.wdl
View File
| task SepSnvIndel { | |||||
| File vcf | |||||
| String sampleName = basename(vcf,".normed.vcf") | |||||
| File keeped_vcf | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| cat ${vcf} | grep '#' > header | |||||
| cat ${vcf} | sed '/^#/d' | awk '$5!~/,/' > removed.body | |||||
| cat ${vcf} | sed '/^#/d' | awk '$5~/,/' > MNP.body | |||||
| cat header removed.body > ${sampleName}.MNPremoved.vcf | |||||
| cat header MNP.body > ${sampleName}.MNP.vcf | |||||
| rtg bgzip ${sampleName}.MNPremoved.vcf | |||||
| rtg index -f vcf ${sampleName}.MNPremoved.vcf.gz | |||||
| rtg bgzip ${keeped_vcf} -c > all.selected.vcf.gz | |||||
| rtg index -f vcf all.selected.vcf.gz | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.snv.train.vcf.gz --include-vcf=all.selected.vcf.gz --snps-only | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.snv.test.vcf.gz --exclude-vcf=all.selected.vcf.gz --snps-only | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.indel.train.vcf.gz --include-vcf=all.selected.vcf.gz --non-snps-only | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.indel.test.vcf.gz --exclude-vcf=all.selected.vcf.gz --non-snps-only | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.snv.vcf.gz --snps-only | |||||
| rtg vcffilter -i ${sampleName}.MNPremoved.vcf.gz -o ${sampleName}.normed.indel.vcf.gz --non-snps-only | |||||
| gzip -d ${sampleName}.normed.snv.train.vcf.gz -c > ${sampleName}.normed.snv.train.vcf | |||||
| gzip -d ${sampleName}.normed.snv.test.vcf.gz -c > ${sampleName}.normed.snv.test.vcf | |||||
| gzip -d ${sampleName}.normed.indel.train.vcf.gz -c > ${sampleName}.normed.indel.train.vcf | |||||
| gzip -d ${sampleName}.normed.indel.test.vcf.gz -c > ${sampleName}.normed.indel.test.vcf | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File MNP="${sampleName}.MNP.vcf" | |||||
| File snv_gz = "${sampleName}.normed.snv.vcf.gz" | |||||
| File snv_idx = "${sampleName}.normed.snv.vcf.gz.tbi" | |||||
| File indel_gz = "${sampleName}.normed.indel.vcf.gz" | |||||
| File indel_idx = "${sampleName}.normed.indel.vcf.gz.tbi" | |||||
| File snv_train = "${sampleName}.normed.snv.train.vcf" | |||||
| File snv_test = "${sampleName}.normed.snv.test.vcf" | |||||
| File indel_train = "${sampleName}.normed.indel.train.vcf" | |||||
| File indel_test = "${sampleName}.normed.indel.test.vcf" | |||||
| File snv_train_gz = "${sampleName}.normed.snv.train.vcf.gz" | |||||
| File snv_test_gz = "${sampleName}.normed.snv.test.vcf.gz" | |||||
| File indel_train_gz = "${sampleName}.normed.indel.train.vcf.gz" | |||||
| File indel_test_gz = "${sampleName}.normed.indel.test.vcf.gz" | |||||
| File snv_train_idx = "${sampleName}.normed.snv.train.vcf.gz.tbi" | |||||
| File snv_test_idx = "${sampleName}.normed.snv.test.vcf.gz.tbi" | |||||
| File indel_train_idx = "${sampleName}.normed.indel.train.vcf.gz.tbi" | |||||
| File indel_test_idx = "${sampleName}.normed.indel.test.vcf.gz.tbi" | |||||
| } | |||||
| } |
+ 71
- 0
tasks/SepTrueFalse.wdl
View File
| task SepTrueFalse { | |||||
| File snv_true_bed | |||||
| File snv_false_bed | |||||
| File indel_true_bed | |||||
| File indel_false_bed | |||||
| File snv_padding | |||||
| File indel_padding | |||||
| File snv_gz | |||||
| File indel_gz | |||||
| File snv_idx | |||||
| File indel_idx | |||||
| File snv_test_gz | |||||
| File indel_test_gz | |||||
| File snv_test_idx | |||||
| File indel_test_idx | |||||
| String sampleName = basename(snv_gz,".normed.snv.vcf.gz") | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg vcffilter -i ${snv_test_gz} -o ${sampleName}.true.snv.vcf.gz --include-bed=${snv_true_bed} | |||||
| rtg vcffilter -i ${snv_test_gz} -o ${sampleName}.false.snv.vcf.gz --include-bed=${snv_false_bed} | |||||
| rtg vcffilter -i ${snv_gz} -o ${sampleName}.remain.snv.vcf.gz --exclude-bed=${snv_false_bed} | |||||
| rtg vcffilter -i ${snv_gz} -o ${sampleName}.padding.snv.vcf.gz --include-bed=${snv_padding} | |||||
| rtg vcffilter -i ${indel_test_gz} -o ${sampleName}.true.indel.vcf.gz --include-bed=${indel_true_bed} | |||||
| rtg vcffilter -i ${indel_test_gz} -o ${sampleName}.false.indel.vcf.gz --include-bed=${indel_false_bed} | |||||
| rtg vcffilter -i ${indel_gz} -o ${sampleName}.remain.indel.vcf.gz --exclude-bed=${indel_false_bed} | |||||
| rtg vcffilter -i ${indel_gz} -o ${sampleName}.padding.indel.vcf.gz --include-bed=${indel_padding} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File snv_true_vcf = "${sampleName}.true.snv.vcf.gz" | |||||
| File snv_true_vcf_index = "${sampleName}.true.snv.vcf.gz.tbi" | |||||
| File snv_false_vcf = "${sampleName}.false.snv.vcf.gz" | |||||
| File snv_false_vcf_index = "${sampleName}.false.snv.vcf.gz.tbi" | |||||
| File snv_remain_vcf = "${sampleName}.remain.snv.vcf.gz" | |||||
| File snv_remain_vcf_index = "${sampleName}.remain.snv.vcf.gz.tbi" | |||||
| File snv_padding_vcf = "${sampleName}.padding.snv.vcf.gz" | |||||
| File snv_padding_vcf_index = "${sampleName}.padding.snv.vcf.gz.tbi" | |||||
| File indel_true_vcf = "${sampleName}.true.indel.vcf.gz" | |||||
| File indel_true_vcf_index = "${sampleName}.true.indel.vcf.gz.tbi" | |||||
| File indel_false_vcf = "${sampleName}.false.indel.vcf.gz" | |||||
| File indel_false_vcf_index = "${sampleName}.false.indel.vcf.gz.tbi" | |||||
| File indel_remain_vcf = "${sampleName}.remain.indel.vcf.gz" | |||||
| File indel_remain_vcf_index = "${sampleName}.remain.indel.vcf.gz.tbi" | |||||
| File indel_padding_vcf = "${sampleName}.padding.indel.vcf.gz" | |||||
| File indel_padding_vcf_index = "${sampleName}.padding.indel.vcf.gz.tbi" | |||||
| } | |||||
| } | |||||
+ 60
- 0
tasks/mendelian.wdl
View File
| task mendelian { | |||||
| File LCL5_vcf | |||||
| File LCL6_vcf | |||||
| File LCL7_vcf | |||||
| File LCL8_vcf | |||||
| String LCL5_name | |||||
| String LCL6_name | |||||
| String LCL7_name | |||||
| String LCL8_name | |||||
| String family_name | |||||
| File ref_dir | |||||
| String fasta | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||||
| mkdir sister | |||||
| /opt/VBT-TrioAnalysis/vbt varcomp -called ${LCL5_vcf} -base ${LCL6_vcf} -ref ${ref_dir}/${fasta} -outDir sister -filter none | |||||
| mv sister/TPBase.vcf ${family_name}.sister.consistent.vcf | |||||
| mv sister/FP.vcf ${family_name}.LCL5.uniq.vcf | |||||
| mv sister/FN.vcf ${family_name}.LCL6.uniq.vcf | |||||
| mv sister/log.txt ${family_name}.sister.vbt.log.txt | |||||
| mkdir VBT | |||||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${LCL8_vcf} -father ${LCL7_vcf} -child ${family_name}.sister.consistent.vcf -outDir VBT -out-prefix ${family_name} --output-violation-regions | |||||
| cat VBT/${family_name}_trio.vcf | grep '#' | cut -f1-9,10 > mother_header | |||||
| cat VBT/${family_name}_trio.vcf | grep -v '#' | cut -f1-9,10 | grep 'MD=1' | grep -v '0/0' | cat mother_header - > ${LCL8_name}.sister.mendelian.gt.vcf | |||||
| cat VBT/${family_name}_trio.vcf | grep '#' | cut -f1-9,11 > father_header | |||||
| cat VBT/${family_name}_trio.vcf | grep -v '#' | cut -f1-9,11 | grep 'MD=1' | grep -v '0/0' | cat father_header - > ${LCL7_name}.sister.mendelian.gt.vcf | |||||
| cat VBT/${family_name}_trio.vcf | grep '#' | cut -f1-9,12 > twin_header | |||||
| cat VBT/${family_name}_trio.vcf | grep -v '#' | cut -f1-9,12 | grep 'MD=1' | grep -v '0/0' | cat header - > ${family_name}.twins.sister.mendelian.gt.vcf | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File sister_consistent_vcf = "${family_name}.sister.consistent.vcf" | |||||
| File LCL5_uniq_vcf = "${family_name}.LCL5.uniq.vcf" | |||||
| File LCL6_uniq_vcf = "${family_name}.LCL6.uniq.vcf" | |||||
| File sister_log = "${family_name}.sister.vbt.log.txt" | |||||
| Array[File] vbt_mendelian = glob("VBT/*") | |||||
| File mother_vcf = "${LCL8_name}.sister.mendelian.gt.vcf" | |||||
| File father_vcf = "${LCL7_name}.sister.mendelian.gt.vcf" | |||||
| File twins_vcf = "${family_name}.twins.sister.mendelian.gt.vcf" | |||||
| } | |||||
| } |
+ 31
- 0
tasks/merge.wdl
View File
| task merge { | |||||
| Array[File] mother_vcf_gz | |||||
| Array[File] mother_vcf_idx | |||||
| Array[File] father_vcf_gz | |||||
| Array[File] father_vcf_idx | |||||
| Array[File] twins_vcf_gz | |||||
| Array[File] twins_vcf_idx | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o LCL8.sister.consistent.merged.vcf ${sep=" " mother_vcf_gz} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o LCL7.sister.consistent.merged.vcf ${sep=" " father_vcf_gz} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o Twins.sister.consistent.vcf ${sep=" " twins_vcf_gz} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File mother_merged_vcf = "LCL8.sister.consistent.merged.vcf" | |||||
| File father_merged_vcf = "LCL7.sister.consistent.merged.vcf" | |||||
| File twins_merged_vcf = "Twins.sister.consistent.merged.vcf" | |||||
| } | |||||
| } |
+ 35
- 0
tasks/mergeBed.wdl
View File
| task mergeBed { | |||||
| Array[File] snv_true_bed | |||||
| Array[File] snv_false_bed | |||||
| Array[File] indel_true_bed | |||||
| Array[File] indel_false_bed | |||||
| Array[File] indel_padding | |||||
| Array[File] snv_padding | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools multiinter -i ${sep=" " snv_true_bed} ${sep=" " indel_true_bed} > merged.true.bed | |||||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools multiinter -i ${sep=" " snv_false_bed} ${sep=" " indel_false_bed} > merged.false.bed | |||||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools multiinter -i ${sep=" " snv_padding} ${sep=" " indel_padding} > merged.padding.bed | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File true_bed = "merged.true.bed" | |||||
| File false_bed = "merged.false.bed" | |||||
| File padding = "merged.padding.bed" | |||||
| } | |||||
| } | |||||
+ 61
- 0
tasks/mergeVCF.wdl
View File
| task mergeVCF { | |||||
| Array[File] snv_true_vcf | |||||
| Array[File] snv_true_vcf_index | |||||
| Array[File] snv_false_vcf | |||||
| Array[File] snv_false_vcf_index | |||||
| Array[File] snv_remain_vcf | |||||
| Array[File] snv_remain_vcf_index | |||||
| Array[File] snv_padding_vcf | |||||
| Array[File] snv_padding_vcf_index | |||||
| Array[File] indel_true_vcf | |||||
| Array[File] indel_true_vcf_index | |||||
| Array[File] indel_false_vcf | |||||
| Array[File] indel_false_vcf_index | |||||
| Array[File] indel_remain_vcf | |||||
| Array[File] indel_remain_vcf_index | |||||
| Array[File] indel_padding_vcf | |||||
| Array[File] indel_padding_vcf_index | |||||
| String quartet_sample | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.snv.true.vcf.gz ${sep=" " snv_true_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.snv.false.vcf.gz ${sep=" " snv_false_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.snv.remain.vcf.gz ${sep=" " snv_remain_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.snv.padding.vcf.gz ${sep=" " snv_padding_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.indel.true.vcf.gz ${sep=" " indel_true_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.indel.false.vcf.gz ${sep=" " indel_false_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.indel.remain.vcf.gz ${sep=" " indel_remain_vcf} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o ${quartet_sample}.indel.padding.vcf.gz ${sep=" " indel_padding_vcf} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File merged_snv_true = "${quartet_sample}.snv.true.vcf.gz" | |||||
| File merged_snv_false = "${quartet_sample}.snv.false.vcf.gz" | |||||
| File merged_snv_remain = "${quartet_sample}.snv.remain.vcf.gz" | |||||
| File merged_snv_padding = "${quartet_sample}.snv.padding.vcf.gz" | |||||
| File merged_indel_true = "${quartet_sample}.indel.true.vcf.gz" | |||||
| File merged_indel_false = "${quartet_sample}.indel.false.vcf.gz" | |||||
| File merged_indel_remain = "${quartet_sample}.indel.remain.vcf.gz" | |||||
| File merged_indel_padding = "${quartet_sample}.indel.padding.vcf.gz" | |||||
| } | |||||
| } |
+ 39
- 0
tasks/oneClass.wdl
View File
| task oneClass { | |||||
| File snv_train_vcf | |||||
| File snv_test_vcf | |||||
| File indel_train_vcf | |||||
| File indel_test_vcf | |||||
| String sampleName = basename(snv_train_vcf,".normed.snv.train.txt") | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| python /opt/oneClass.py -train ${snv_train_vcf} -test ${snv_test_vcf} -name ${sampleName}_snv | |||||
| python /opt/oneClass.py -train ${indel_train_vcf} -test ${indel_test_vcf} -name ${sampleName}_indel | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File snv_true_txt = "${sampleName}_snv_predicted_true.txt" | |||||
| File snv_false_txt = "${sampleName}_snv_predicted_false.txt" | |||||
| File snv_true_bed = "${sampleName}_snv_predicted_true.bed" | |||||
| File snv_false_bed = "${sampleName}_snv_predicted_false.bed" | |||||
| File snv_padding = "${sampleName}_snv_padding.bed" | |||||
| File indel_true_txt = "${sampleName}_indel_predicted_true.txt" | |||||
| File indel_false_txt = "${sampleName}_indel_predicted_false.txt" | |||||
| File indel_true_bed = "${sampleName}_indel_predicted_true.bed" | |||||
| File indel_false_bed = "${sampleName}_indel_predicted_false.bed" | |||||
| File indel_padding = "${sampleName}_indel_padding.bed" | |||||
| } | |||||
| } | |||||
+ 33
- 0
tasks/sister.wdl
View File
| task sister { | |||||
| File LCL5_vcf | |||||
| File LCL6_vcf | |||||
| File ref_dir | |||||
| String fasta | |||||
| String family_name | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| mkdir sister | |||||
| vbt varcomp -called ${LCL5_vcf} -base ${LCL6_vcf} -ref ${ref_dir}/${fasta} -outDir sister -filter none | |||||
| mv sister/TPBase.vcf ${family_name}.sister.consistent.vcf | |||||
| mv sister/FP.vcf ${family_name}.LCL5.uniq.vcf | |||||
| mv sister/FN.vcf ${family_name}.LCL6.uniq.vcf | |||||
| mv sister/log.txt ${family_name}.sister.vbt.log.txt | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File sister_consistent_vcf = "${family_name}.sister.consistent.vcf" | |||||
| File LCL5_uniq = "${family_name}.LCL5.uniq.vcf" | |||||
| File LCL6_uniq = "${family_name}.LCL6.uniq.vcf" | |||||
| File log = "${family_name}.sister.vbt.log.txt" | |||||
| } | |||||
| } |
+ 30
- 0
tasks/variantsNorm.wdl
View File
| task variantsNorm { | |||||
| File vcf | |||||
| File ref_dir | |||||
| String fasta | |||||
| String sampleName | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| cat ${vcf} | grep '#' > header | |||||
| cat ${vcf} | grep -v '#' > body | |||||
| cat body | grep -w '^chr1\|^chr2\|^chr3\|^chr4\|^chr5\|^chr6\|^chr7\|^chr8\|^chr9\|^chr10\|^chr11\|^chr12\|^chr13\|^chr14\|^chr15\|^chr16\|^chr17\|^chr18\|^chr19\|^chr20\|^chr21\|^chr22\|^chrX' > body.filtered | |||||
| cat header body.filtered > ${sampleName}.filtered.vcf | |||||
| /opt/hall-lab/bcftools-1.9/bin/bcftools norm -f ${ref_dir}/${fasta} ${sampleName}.filtered.vcf > ${sampleName}.normed.vcf | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File normed_vcf = "${sampleName}.normed.vcf" | |||||
| } | |||||
| } |
+ 31
- 0
tasks/votes.wdl
View File
| task votes { | |||||
| Array[File] mother_vcf_gz | |||||
| Array[File] mother_vcf_idx | |||||
| Array[File] father_vcf_gz | |||||
| Array[File] father_vcf_idx | |||||
| Array[File] twins_vcf_gz | |||||
| Array[File] twins_vcf_idx | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o LCL8.sister.consistent.merged.vcf ${sep=" " mother_vcf_gz} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o LCL7.sister.consistent.merged.vcf ${sep=" " father_vcf_gz} | |||||
| rtg vcfmerge --force-merge-all --no-gzip -o Twins.sister.consistent.vcf ${sep=" " twins_vcf_gz} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File mother_merged_vcf = "LCL8.sister.consistent.merged.vcf" | |||||
| File father_merged_vcf = "LCL7.sister.consistent.merged.vcf" | |||||
| File twins_merged_vcf = "Twins.sister.consistent.merged.vcf" | |||||
| } | |||||
| } |
+ 35
- 0
tasks/zipIndex.wdl
View File
| task zipIndex { | |||||
| File mother_vcf | |||||
| File father_vcf | |||||
| File twins_vcf | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| rtg bgzip ${mother_vcf} | |||||
| rtg index -f vcf ${mother_vcf}.gz | |||||
| rtg bgzip ${father_vcf} | |||||
| rtg index -f vcf ${father_vcf}.gz | |||||
| rtg bgzip ${twins_vcf} | |||||
| rtg index -f vcf ${twins_vcf}.gz | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File mother_vcf_gz = "${mother_vcf}.gz" | |||||
| File father_vcf_gz = "${father_vcf}.gz" | |||||
| File twins_vcf_gz = "${twins_vcf}.gz" | |||||
| File mother_vcf_idx = "${mother_vcf}.gz.tbi" | |||||
| File father_vcf_idx = "${father_vcf}.gz.tbi" | |||||
| File twins_vcf_idx = "${twins_vcf}.gz.tbi" | |||||
| } | |||||
| } |
+ 88
- 0
workflow.wdl
View File
| import "./tasks/variantsNorm.wdl" as variantsNorm | |||||
| import "./tasks/mendelian.wdl" as mendelian | |||||
| import "./tasks/zipIndex.wdl" as zipIndex | |||||
| import "./tasks/merge.wdl" as merge | |||||
| workflow {{ project_name }} { | |||||
| File inputSamplesFile | |||||
| Array[Array[File]] inputSamples = read_tsv(inputSamplesFile) | |||||
| File ref_dir | |||||
| String fasta | |||||
| String cluster_config | |||||
| String disk_size | |||||
| scatter (sample in inputSamples){ | |||||
| call variantsNorm.variantsNorm as LCL5variantsNorm{ | |||||
| input: | |||||
| vcf=sample[0], | |||||
| ref_dir=ref_dir, | |||||
| fasta=fasta, | |||||
| sampleName=sample[4], | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| call variantsNorm.variantsNorm as LCL6variantsNorm{ | |||||
| input: | |||||
| vcf=sample[1], | |||||
| ref_dir=ref_dir, | |||||
| fasta=fasta, | |||||
| sampleName=sample[5], | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| call variantsNorm.variantsNorm as LCL7variantsNorm{ | |||||
| input: | |||||
| vcf=sample[2], | |||||
| ref_dir=ref_dir, | |||||
| fasta=fasta, | |||||
| sampleName=sample[6], | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| call variantsNorm.variantsNorm as LCL8variantsNorm{ | |||||
| input: | |||||
| vcf=sample[3], | |||||
| ref_dir=ref_dir, | |||||
| fasta=fasta, | |||||
| sampleName=sample[7], | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| call mendelian.mendelian as mendelian { | |||||
| input: | |||||
| LCL5_vcf=LCL5variantsNorm.normed_vcf, | |||||
| LCL6_vcf=LCL6variantsNorm.normed_vcf, | |||||
| LCL7_vcf=LCL7variantsNorm.normed_vcf, | |||||
| LCL8_vcf=LCL8variantsNorm.normed_vcf, | |||||
| LCL5_name=sample[4], | |||||
| LCL6_name=sample[5], | |||||
| LCL7_name=sample[6], | |||||
| LCL8_name=sample[7], | |||||
| family_name=sample[8], | |||||
| ref_dir=ref_dir, | |||||
| fasta=fasta, | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| call zipIndex.zipIndex as zipIndex{ | |||||
| input: | |||||
| mother_vcf=mendelian.mother_vcf, | |||||
| father_vcf=mendelian.father_vcf, | |||||
| twins_vcf=mendelian.twins_vcf, | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| } | |||||
| call merge.merge as merge { | |||||
| input: | |||||
| mother_vcf_gz=zipIndex.mother_vcf_gz, | |||||
| mother_vcf_idx=zipIndex.mother_vcf_idx, | |||||
| father_vcf_gz=zipIndex.father_vcf_gz, | |||||
| father_vcf_idx=zipIndex.father_vcf_idx, | |||||
| twins_vcf_gz=zipIndex.twins_vcf_gz, | |||||
| twins_vcf_idx=zipIndex.twins_vcf_idx, | |||||
| cluster_config=cluster_config, | |||||
| disk_size=disk_size | |||||
| } | |||||
| } |
Loading…