
 LUYAO REN
5年前
LUYAO REN
5年前
コミット
3bdfa208f1
29個のファイルの変更、1621行の追加、0行の削除
+ 103
- 0
README.md
ファイルの表示
| @@ -0,0 +1,103 @@ | |||
| #高置信突变位点的整合 | |||
| > Author: Run Luyao | |||
| > | |||
| > E-mail:18110700050@fudan.edu.cn | |||
| > | |||
| > Git:http://choppy.3steps.cn/renluyao/high_confidence_calls_manuscript.git | |||
| > | |||
| > Last Updates: 18/03/2020 | |||
| ## 安装指南 | |||
| ```bash | |||
| # 激活choppy环境 | |||
| source activate choppy | |||
| # 安装app | |||
| choppy install renluyao/high_confidence_calls_manuscript | |||
| ``` | |||
| ## App概述 | |||
| 中华家系1号全基因组高置信small variants(SNVs和Indels)的整合流程。 | |||
| ## 流程与参数 | |||
| ####1. variantsNorm | |||
| 保留chr1-22,X上的突变 | |||
| 用bcftools norm进行突变格式的统一 | |||
| ####2. mendelian | |||
| LCL5、LCL7和LCL8为三口之家,进行trio-analysis,分析符合孟德尔和不符合孟德尔遗传规律的突变位点 | |||
| LCL6、LCL7和LCL8为三口之家,进行trio-analysis,分析符合孟德尔和不符合孟德尔遗传规律的突变位点 | |||
| 得到LCL5和LCL6两个家系合并的vcf文件 | |||
| ####3. zipIndex | |||
| 对LCL5和LCL6两个家系合并的文件压缩和检索引 | |||
| ####4. VCFrename | |||
| 将VBT的输出结果,VCF文件中的MOTHER FATHER CHILD改成对应的样本名 | |||
| ####5. mergeSister | |||
| 将LCL5和LCL6修改过名字后的家系VCF文件合并 | |||
| ####6. reformVCF | |||
| 根据两个三口之家和姐妹的孟德尔遗传的信息,将之前和合并的VCF分解成4个人的vcf,并且包含了家系遗传的信息 | |||
| ####7. familyzipIndex | |||
| 将上一步输出的4个文件进行压缩和检索引 | |||
| ####8. merge | |||
| 将所有注释后的LCL5 vcf文件合并 | |||
| 将所有注释后的LCL6 vcf文件合并 | |||
| 将所有注释后的LCL7 vcf文件合并 | |||
| 将所有注释后的LCL8 vcf文件合并 | |||
| ####9. vote | |||
| 投票选择高置信的突变位点 | |||
| t | |||
| ## App输入变量与输入文件 | |||
| inputSamplesFile的格式如下 | |||
| ```bash | |||
| #LCL5_VCF #LCL6_VCF #LCL7_VCF #LCL8_VCF #LCL5_sampleName #LCL6_sampleName #LCL7_sampleName #LCL8_sampleName #familyName | |||
| ``` | |||
| 最终版的整合文件包括: | |||
| ## App输出文件 | |||
| ## 结果展示与解读 | |||
| ## CHANGELOG | |||
| ## FAQ | |||
バイナリ
codescripts/.DS_Store
ファイルの表示
+ 92
- 0
codescripts/extract_vcf_information.py
ファイルの表示
| @@ -0,0 +1,92 @@ | |||
| import sys,getopt | |||
| import os | |||
| import re | |||
| import fileinput | |||
| import pandas as pd | |||
| def usage(): | |||
| print( | |||
| """ | |||
| Usage: python extract_vcf_information.py -i input_merged_vcf_file -o parsed_file | |||
| This script will extract SNVs and Indels information from the vcf files and output a tab-delimited files. | |||
| Input: | |||
| -i the selected vcf file | |||
| Output: | |||
| -o tab-delimited parsed file | |||
| """) | |||
| # select supported small variants | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| infoParsed = parse_INFO(strings[7]) | |||
| formatKeys = strings[8].split(':') | |||
| formatValues = strings[9].split(':') | |||
| for i in range(0,len(formatKeys) -1) : | |||
| if formatKeys[i] == 'AD': | |||
| ra = formatValues[i].split(',') | |||
| infoParsed['RefDP'] = ra[0] | |||
| infoParsed['AltDP'] = ra[1] | |||
| if (int(ra[1]) + int(ra[0])) != 0: | |||
| infoParsed['af'] = float(int(ra[1])/(int(ra[1]) + int(ra[0]))) | |||
| else: | |||
| pass | |||
| else: | |||
| infoParsed[formatKeys[i]] = formatValues[i] | |||
| infoParsed['chromo'] = strings[0] | |||
| infoParsed['pos'] = strings[1] | |||
| infoParsed['id'] = strings[2] | |||
| infoParsed['ref'] = strings[3] | |||
| infoParsed['alt'] = strings[4] | |||
| infoParsed['qual'] = strings[5] | |||
| return infoParsed | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| elif kv[0] == 'AF': | |||
| pass | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||
| for op,value in opts: | |||
| if op == "-i": | |||
| inputFile=value | |||
| elif op == "-o": | |||
| outputFile=value | |||
| elif op == "-h": | |||
| usage() | |||
| sys.exit() | |||
| if len(sys.argv[1:]) < 3: | |||
| usage() | |||
| sys.exit() | |||
| allDict = [] | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| oneDict = process(line) | |||
| allDict.append(oneDict) | |||
| allTable = pd.DataFrame(allDict) | |||
| allTable.to_csv(outputFile,sep='\t',index=False) | |||
+ 416
- 0
codescripts/high_confidence_call_vote.py
ファイルの表示
| @@ -0,0 +1,416 @@ | |||
| from __future__ import division | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to count voting number") | |||
| parser.add_argument('-vcf', '--multi_sample_vcf', type=str, help='The VCF file you want to count the voting number', required=True) | |||
| parser.add_argument('-dup', '--dup_list', type=str, help='Duplication list', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='Prefix of output file name', required=True) | |||
| args = parser.parse_args() | |||
| multi_sample_vcf = args.multi_sample_vcf | |||
| dup_list = args.dup_list | |||
| prefix = args.prefix | |||
| sample_name = args.sample_name | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Sum depth of all samples"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Sum alternative depth of all samples"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency, sum alternative depth / sum depth"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Average genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Average mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUALI,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| vcf_header_all_sample = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DUP,Number=1,Type=Flag,Description="Duplicated variant records"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##INFO=<ID=ALL_ALT,Number=1,Type=Float,Description="Sum of alternative reads of all samples"> | |||
| ##INFO=<ID=ALL_DP,Number=1,Type=Float,Description="Sum of depth of all samples"> | |||
| ##INFO=<ID=ALL_AF,Number=1,Type=Float,Description="Allele frequency of net alternatice reads and net depth"> | |||
| ##INFO=<ID=GQ_MEAN,Number=1,Type=Float,Description="Mean of genotype quality of all samples"> | |||
| ##INFO=<ID=QD_MEAN,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##INFO=<ID=MQ_MEAN,Number=1,Type=Float,Description="Mean of mapping quality of all samples"> | |||
| ##INFO=<ID=FS_MEAN,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##INFO=<ID=QUAL_MEAN,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##INFO=<ID=PCR,Number=1,Type=String,Description="Consensus of PCR votes"> | |||
| ##INFO=<ID=PCR_FREE,Number=1,Type=String,Description="Consensus of PCR-free votes"> | |||
| ##INFO=<ID=CONSENSUS,Number=1,Type=String,Description="Consensus calls"> | |||
| ##INFO=<ID=CONSENSUS_SEQ,Number=1,Type=String,Description="Consensus sequence"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=String,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=String,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=String,Description="Genotype quality"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=String,Description="Mapping quality"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=String,Description="1 is twins shared, 0 is twins discordant "> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL5 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL6 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # read in duplication list | |||
| dup = pd.read_table(dup_list,header=None) | |||
| var_dup = dup[0].tolist() | |||
| # output file | |||
| benchmark_file_name = prefix + '_voted.vcf' | |||
| benchmark_outfile = open(benchmark_file_name,'w') | |||
| all_sample_file_name = prefix + '_all_sample_information.vcf' | |||
| all_sample_outfile = open(all_sample_file_name,'w') | |||
| # write VCF | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '_benchmark_calls\n' | |||
| benchmark_outfile.write(vcf_header) | |||
| benchmark_outfile.write(outputcolumn) | |||
| outputcolumn_all_sample = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+ \ | |||
| 'Quartet_DNA_BGI_SEQ2000_BGI_1_20180518\tQuartet_DNA_BGI_SEQ2000_BGI_2_20180530\tQuartet_DNA_BGI_SEQ2000_BGI_3_20180530\t' + \ | |||
| 'Quartet_DNA_BGI_T7_WGE_1_20191105\tQuartet_DNA_BGI_T7_WGE_2_20191105\tQuartet_DNA_BGI_T7_WGE_3_20191105\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_1_20181108\tQuartet_DNA_ILM_Nova_ARD_2_20181108\tQuartet_DNA_ILM_Nova_ARD_3_20181108\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_4_20190111\tQuartet_DNA_ILM_Nova_ARD_5_20190111\tQuartet_DNA_ILM_Nova_ARD_6_20190111\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_BRG_1_20180930\tQuartet_DNA_ILM_Nova_BRG_2_20180930\tQuartet_DNA_ILM_Nova_BRG_3_20180930\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_WUX_1_20190917\tQuartet_DNA_ILM_Nova_WUX_2_20190917\tQuartet_DNA_ILM_Nova_WUX_3_20190917\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_ARD_1_20170403\tQuartet_DNA_ILM_XTen_ARD_2_20170403\tQuartet_DNA_ILM_XTen_ARD_3_20170403\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_NVG_1_20170329\tQuartet_DNA_ILM_XTen_NVG_2_20170329\tQuartet_DNA_ILM_XTen_NVG_3_20170329\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_WUX_1_20170216\tQuartet_DNA_ILM_XTen_WUX_2_20170216\tQuartet_DNA_ILM_XTen_WUX_3_20170216\n' | |||
| all_sample_outfile.write(vcf_header_all_sample) | |||
| all_sample_outfile.write(outputcolumn_all_sample) | |||
| #function | |||
| def replace_nan(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| updated_list.append('.:.:.:.:.:.:.:.:.:.:.:.') | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def remove_dot(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| pass | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def detected_number(strings): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| percentage = 27 - gt.count('.') | |||
| return(str(percentage)) | |||
| def vote_number(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| vote_num = gt.count(consensus_call) | |||
| return(str(vote_num)) | |||
| def family_vote(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| indices = [i for i, x in enumerate(gt) if x == consensus_call] | |||
| matched_mendelian = itemgetter(*indices)(mendelian) | |||
| mendelian_num = matched_mendelian.count('1:1:1') | |||
| return(str(mendelian_num)) | |||
| def gt_uniform(strings): | |||
| uniformed_gt = '' | |||
| allele1 = strings.split('/')[0] | |||
| allele2 = strings.split('/')[1] | |||
| if int(allele1) > int(allele2): | |||
| uniformed_gt = allele2 + '/' + allele1 | |||
| else: | |||
| uniformed_gt = allele1 + '/' + allele2 | |||
| return uniformed_gt | |||
| def decide_by_rep(strings): | |||
| consensus_rep = '' | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| # modified gt turn 2/1 to 1/2 | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| # mendelian consistent? | |||
| mendelian_dict = Counter(mendelian) | |||
| highest_mendelian = mendelian_dict.most_common(1) | |||
| candidate_mendelian = highest_mendelian[0][0] | |||
| freq_mendelian = highest_mendelian[0][1] | |||
| if (candidate_mendelian == '1:1:1') and (freq_mendelian >= 2): | |||
| gt_num_dict = Counter(gt) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if (candidate_gt != '0/0') and (freq_gt >= 2): | |||
| consensus_rep = candidate_gt | |||
| elif (candidate_gt == '0/0') and (freq_gt >= 2): | |||
| consensus_rep = '0/0' | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| elif (candidate_mendelian == '') and (freq_mendelian >= 2): | |||
| consensus_rep = 'noInfo' | |||
| else: | |||
| consensus_rep = 'inconMen' | |||
| return consensus_rep | |||
| def main(): | |||
| for line in fileinput.input(multi_sample_vcf): | |||
| headline = re.match('^\#',line) | |||
| if headline is not None: | |||
| pass | |||
| else: | |||
| line = line.strip() | |||
| strings = line.split('\t') | |||
| variant_id = '_'.join([strings[0],strings[1]]) | |||
| # check if the variants location is duplicated | |||
| if variant_id in var_dup: | |||
| strings[7] = strings[7] + ';DUP' | |||
| outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(outLine) | |||
| else: | |||
| # pre-define | |||
| pcr_consensus = '.' | |||
| pcr_free_consensus = '.' | |||
| consensus_call = '.' | |||
| consensus_alt_seq = '.' | |||
| # pcr | |||
| strings[9:] = replace_nan(strings[9:]) | |||
| pcr = itemgetter(*[9,10,11,27,28,29,30,31,32,33,34,35])(strings) | |||
| SEQ2000 = decide_by_rep(pcr[0:3]) | |||
| XTen_ARD = decide_by_rep(pcr[3:6]) | |||
| XTen_NVG = decide_by_rep(pcr[6:9]) | |||
| XTen_WUX = decide_by_rep(pcr[9:12]) | |||
| sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 2: | |||
| pcr_consensus = candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| pcr_free = itemgetter(*[12,13,14,15,16,17,18,19,20,21,22,23,24,25,26])(strings) | |||
| T7_WGE = decide_by_rep(pcr_free[0:3]) | |||
| Nova_ARD_1 = decide_by_rep(pcr_free[3:6]) | |||
| Nova_ARD_2 = decide_by_rep(pcr_free[6:9]) | |||
| Nova_BRG = decide_by_rep(pcr_free[9:12]) | |||
| Nova_WUX = decide_by_rep(pcr_free[12:15]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG,Nova_WUX] | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 3: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| # pcr and pcr-free | |||
| tag = ['inconGT','noInfo','inconMen','inconSequenceSite'] | |||
| if (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag) and (pcr_consensus != '0/0'): | |||
| consensus_call = pcr_consensus | |||
| VOTED = vote_number(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';VOTED=' + VOTED | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| FAM = family_vote(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';FAM=' + FAM | |||
| # Delete multiple alternative genotype to necessary expression | |||
| alt = strings[4] | |||
| alt_gt = alt.split(',') | |||
| if len(alt_gt) > 1: | |||
| allele1 = consensus_call.split('/')[0] | |||
| allele2 = consensus_call.split('/')[1] | |||
| if allele1 == '0': | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| consensus_alt_seq = allele2_seq | |||
| consensus_call = '0/1' | |||
| else: | |||
| allele1_seq = alt_gt[int(allele1) - 1] | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| if int(allele1) > int(allele2): | |||
| consensus_alt_seq = allele2_seq + ',' + allele1_seq | |||
| consensus_call = '1/2' | |||
| elif int(allele1) < int(allele2): | |||
| consensus_alt_seq = allele1_seq + ',' + allele2_seq | |||
| consensus_call = '1/2' | |||
| else: | |||
| consensus_alt_seq = allele1_seq | |||
| consensus_call = '1/1' | |||
| else: | |||
| consensus_alt_seq = alt | |||
| # GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL | |||
| # GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT | |||
| # DP | |||
| DP = [x.split(':')[4] for x in strings[9:]] | |||
| DP = remove_dot(DP) | |||
| DP = [int(x) for x in DP] | |||
| ALL_DP = sum(DP) | |||
| # AF | |||
| ALT = [x.split(':')[5] for x in strings[9:]] | |||
| ALT = remove_dot(ALT) | |||
| ALT = [int(x) for x in ALT] | |||
| ALL_ALT = sum(ALT) | |||
| ALL_AF = round(ALL_ALT/ALL_DP,2) | |||
| # GQ | |||
| GQ = [x.split(':')[7] for x in strings[9:]] | |||
| GQ = remove_dot(GQ) | |||
| GQ = [int(x) for x in GQ] | |||
| GQ_MEAN = round(mean(GQ),2) | |||
| # QD | |||
| QD = [x.split(':')[8] for x in strings[9:]] | |||
| QD = remove_dot(QD) | |||
| QD = [float(x) for x in QD] | |||
| QD_MEAN = round(mean(QD),2) | |||
| # MQ | |||
| MQ = [x.split(':')[9] for x in strings[9:]] | |||
| MQ = remove_dot(MQ) | |||
| MQ = [float(x) for x in MQ] | |||
| MQ_MEAN = round(mean(MQ),2) | |||
| # FS | |||
| FS = [x.split(':')[10] for x in strings[9:]] | |||
| FS = remove_dot(FS) | |||
| FS = [float(x) for x in FS] | |||
| FS_MEAN = round(mean(FS),2) | |||
| # QUAL | |||
| QUAL = [x.split(':')[11] for x in strings[9:]] | |||
| QUAL = remove_dot(QUAL) | |||
| QUAL = [float(x) for x in QUAL] | |||
| QUAL_MEAN = round(mean(QUAL),2) | |||
| # benchmark output | |||
| output_format = consensus_call + ':' + str(ALL_DP) + ':' + str(ALL_ALT) + ':' + str(ALL_AF) + ':' + str(GQ_MEAN) + ':' + str(QD_MEAN) + ':' + str(MQ_MEAN) + ':' + str(FS_MEAN) + ':' + str(QUAL_MEAN) | |||
| outLine = strings[0] + '\t' + strings[1] + '\t' + strings[2] + '\t' + strings[3] + '\t' + consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' + strings[7] + '\t' + 'GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + output_format + '\n' | |||
| benchmark_outfile.write(outLine) | |||
| # all sample output | |||
| strings[7] = strings[7] + ';ALL_ALT=' + str(ALL_ALT) + ';ALL_DP=' + str(ALL_DP) + ';ALL_AF=' + str(ALL_AF) \ | |||
| + ';GQ_MEAN=' + str(GQ_MEAN) + ';QD_MEAN=' + str(QD_MEAN) + ';MQ_MEAN=' + str(MQ_MEAN) + ';FS_MEAN=' + str(FS_MEAN) \ | |||
| + ';QUAL_MEAN=' + str(QUAL_MEAN) + ';PCR=' + consensus_call + ';PCR_FREE=' + consensus_call + ';CONSENSUS=' + consensus_call \ | |||
| + ';CONSENSUS_SEQ=' + consensus_alt_seq | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus == '0/0') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != '0/0')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus != '0/0') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == '0/0')): | |||
| consensus_call = 'pcr-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call + ';PCR=' + pcr_consensus + ';PCR_FREE=' + pcr_free_consensus | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = 'confirm for parents' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| else: | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| if __name__ == '__main__': | |||
| main() | |||
+ 129
- 0
codescripts/merge_mendelian_vcfinfo.py
ファイルの表示
| @@ -0,0 +1,129 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to get final high confidence calls and information of all replicates") | |||
| parser.add_argument('-vcfInfo', '--vcfInfo', type=str, help='The txt file of variants information, this file is named as prefix__variant_quality_location.txt', required=True) | |||
| parser.add_argument('-mendelianInfo', '--mendelianInfo', type=str, help='The merged mendelian information of all samples', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| args = parser.parse_args() | |||
| vcfInfo = args.vcfInfo | |||
| mendelianInfo = args.mendelianInfo | |||
| sample_name = args.sample_name | |||
| #GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=Flag,Description="1 for sister consistent, 0 for sister different"> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL5 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL6 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUAL,Number=1,Type=Float,Description="variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # output file | |||
| file_name = sample_name + '_mendelian_vcfInfo.vcf' | |||
| outfile = open(file_name,'w') | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '\n' | |||
| outfile.write(vcf_header) | |||
| outfile.write(outputcolumn) | |||
| # input files | |||
| vcf_info = pd.read_table(vcfInfo) | |||
| mendelian_info = pd.read_table(mendelianInfo) | |||
| merged_df = pd.merge(vcf_info, mendelian_info, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_df = merged_df.fillna('.') | |||
| # | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| # | |||
| for row in merged_df.itertuples(): | |||
| if row[18] != '.': | |||
| # format | |||
| # GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = row[18] + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_y + '\t' + row.ALT_y + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + FORMAT + '\n' | |||
| else: | |||
| rawGT = row[10].split(':') | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = '.:.:.:.' + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) + ':' + rawGT[0] | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_x + '\t' + row.ALT_x + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT' + '\t' + FORMAT + '\n' | |||
| outfile.write(outline) | |||
+ 109
- 0
codescripts/oneClass.py
ファイルの表示
| @@ -0,0 +1,109 @@ | |||
| # import modules | |||
| import numpy as np | |||
| import pandas as pd | |||
| from sklearn import svm | |||
| from sklearn import preprocessing | |||
| import sys, argparse, os | |||
| from vcf2bed import position_to_bed,padding_region | |||
| parser = argparse.ArgumentParser(description="this script is to preform one calss svm on each chromosome") | |||
| parser.add_argument('-train', '--trainDataset', type=str, help='training dataset generated from extracting vcf information part, with mutaitons supported by callsets', required=True) | |||
| parser.add_argument('-test', '--testDataset', type=str, help='testing dataset generated from extracting vcf information part, with mutaitons not called by all callsets', required=True) | |||
| parser.add_argument('-name', '--sampleName', type=str, help='sample name for output file name', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| train_input = args.trainDataset | |||
| test_input = args.testDataset | |||
| sample_name = args.sampleName | |||
| # default columns, which will be included in the included in the calssifier | |||
| chromosome = ['chr1','chr2','chr3','chr4','chr5','chr6','chr7','chr8','chr9','chr10','chr11','chr12','chr13','chr14','chr15' ,'chr16','chr17','chr18','chr19','chr20','chr21','chr22','chrX','chrY'] | |||
| feature_heter_cols = ['AltDP','BaseQRankSum','DB','DP','FS','GQ','MQ','MQRankSum','QD','ReadPosRankSum','RefDP','SOR','af'] | |||
| feature_homo_cols = ['AltDP','DB','DP','FS','GQ','MQ','QD','RefDP','SOR','af'] | |||
| # import datasets sepearate the records with or without BaseQRankSum annotation, etc. | |||
| def load_dat(dat_file_name): | |||
| dat = pd.read_table(dat_file_name) | |||
| dat['DB'] = dat['DB'].fillna(0) | |||
| dat = dat[dat['DP'] != 0] | |||
| dat['af'] = dat['AltDP']/(dat['AltDP'] + dat['RefDP']) | |||
| homo_rows = dat[dat['BaseQRankSum'].isnull()] | |||
| heter_rows = dat[dat['BaseQRankSum'].notnull()] | |||
| return homo_rows,heter_rows | |||
| train_homo,train_heter = load_dat(train_input) | |||
| test_homo,test_heter = load_dat(test_input) | |||
| clf = svm.OneClassSVM(nu=0.05,kernel='rbf', gamma='auto_deprecated',cache_size=500) | |||
| def prepare_dat(train_dat,test_dat,feature_cols,chromo): | |||
| chr_train = train_dat[train_dat['chromo'] == chromo] | |||
| chr_test = test_dat[test_dat['chromo'] == chromo] | |||
| train_dat = chr_train.loc[:,feature_cols] | |||
| test_dat = chr_test.loc[:,feature_cols] | |||
| train_dat_scaled = preprocessing.scale(train_dat) | |||
| test_dat_scaled = preprocessing.scale(test_dat) | |||
| return chr_test,train_dat_scaled,test_dat_scaled | |||
| def oneclass(X_train,X_test,chr_test): | |||
| clf.fit(X_train) | |||
| y_pred_test = clf.predict(X_test) | |||
| test_true_dat = chr_test[y_pred_test == 1] | |||
| test_false_dat = chr_test[y_pred_test == -1] | |||
| return test_true_dat,test_false_dat | |||
| predicted_true = pd.DataFrame(columns=train_homo.columns) | |||
| predicted_false = pd.DataFrame(columns=train_homo.columns) | |||
| for chromo in chromosome: | |||
| # homo datasets | |||
| chr_test_homo,X_train_homo,X_test_homo = prepare_dat(train_homo,test_homo,feature_homo_cols,chromo) | |||
| test_true_homo,test_false_homo = oneclass(X_train_homo,X_test_homo,chr_test_homo) | |||
| predicted_true = predicted_true.append(test_true_homo) | |||
| predicted_false = predicted_false.append(test_false_homo) | |||
| # heter datasets | |||
| chr_test_heter,X_train_heter,X_test_heter = prepare_dat(train_heter,test_heter,feature_heter_cols,chromo) | |||
| test_true_heter,test_false_heter = oneclass(X_train_heter,X_test_heter,chr_test_heter) | |||
| predicted_true = predicted_true.append(test_true_heter) | |||
| predicted_false = predicted_false.append(test_false_heter) | |||
| predicted_true_filename = sample_name + '_predicted_true.txt' | |||
| predicted_false_filename = sample_name + '_predicted_false.txt' | |||
| predicted_true.to_csv(predicted_true_filename,sep='\t',index=False) | |||
| predicted_false.to_csv(predicted_false_filename,sep='\t',index=False) | |||
| # output the bed file and padding bed region 50bp | |||
| predicted_true_bed_filename = sample_name + '_predicted_true.bed' | |||
| predicted_false_bed_filename = sample_name + '_predicted_false.bed' | |||
| padding_filename = sample_name + '_padding.bed' | |||
| predicted_true_bed = open(predicted_true_bed_filename,'w') | |||
| predicted_false_bed = open(predicted_false_bed_filename,'w') | |||
| padding = open(padding_filename,'w') | |||
| # | |||
| for index,row in predicted_false.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_false_bed.write(outline_pos) | |||
| chromo,pad_pos1,pad_pos2,pad_pos3,pad_pos4 = padding_region(chromo,pos1,pos2,50) | |||
| outline_pad_1 = chromo + '\t' + str(pad_pos1) + '\t' + str(pad_pos2) + '\n' | |||
| outline_pad_2 = chromo + '\t' + str(pad_pos3) + '\t' + str(pad_pos4) + '\n' | |||
| padding.write(outline_pad_1) | |||
| padding.write(outline_pad_2) | |||
| for index,row in predicted_true.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_true_bed.write(outline_pos) | |||
+ 144
- 0
codescripts/reformVCF.py
ファイルの表示
| @@ -0,0 +1,144 @@ | |||
| # import modules | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| parser = argparse.ArgumentParser(description="This script is to split samples in VCF files and rewrite to the right style") | |||
| parser.add_argument('-vcf', '--familyVCF', type=str, help='VCF with sister and mendelian infomation', required=True) | |||
| parser.add_argument('-name', '--familyName', type=str, help='Family name of the VCF file', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| inputFile = args.familyVCF | |||
| family_name = args.familyName | |||
| # output filename | |||
| LCL5_name = family_name + '.LCL5.vcf' | |||
| LCL5file = open(LCL5_name,'w') | |||
| LCL6_name = family_name + '.LCL6.vcf' | |||
| LCL6file = open(LCL6_name,'w') | |||
| LCL7_name = family_name + '.LCL7.vcf' | |||
| LCL7file = open(LCL7_name,'w') | |||
| LCL8_name = family_name + '.LCL8.vcf' | |||
| LCL8file = open(LCL8_name,'w') | |||
| family_filename = family_name + '.vcf' | |||
| familyfile = open(family_filename,'w') | |||
| # default columns, which will be included in the included in the calssifier | |||
| vcfheader = '''##fileformat=VCFv4.2 | |||
| ##FILTER=<ID=PASS,Description="the same genotype between twin sister and mendelian consistent in 578 and 678"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=0,Type=Flag,Description="0 for sister consistent, 1 for sister inconsistent"> | |||
| ##FORMAT=<ID=TRIO5,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##FORMAT=<ID=TRIO6,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # write VCF | |||
| LCL5colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL5'+'\n' | |||
| LCL5file.write(vcfheader) | |||
| LCL5file.write(LCL5colname) | |||
| LCL6colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL6'+'\n' | |||
| LCL6file.write(vcfheader) | |||
| LCL6file.write(LCL6colname) | |||
| LCL7colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL7'+'\n' | |||
| LCL7file.write(vcfheader) | |||
| LCL7file.write(LCL7colname) | |||
| LCL8colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL8'+'\n' | |||
| LCL8file.write(vcfheader) | |||
| LCL8file.write(LCL8colname) | |||
| familycolname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+'LCL5\t'+'LCL6\t'+'LCL7\t'+'LCL8'+'\n' | |||
| familyfile.write(vcfheader) | |||
| familyfile.write(familycolname) | |||
| # reform VCF | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| # replace . | |||
| # LCL5 uniq | |||
| if strings[11] == '.': | |||
| strings[11] = '0/0' | |||
| strings[9] = strings[12] | |||
| strings[10] = strings[13] | |||
| else: | |||
| pass | |||
| # LCL6 uniq | |||
| if strings[14] == '.': | |||
| strings[14] = '0/0' | |||
| strings[12] = strings[9] | |||
| strings[13] = strings[10] | |||
| else: | |||
| pass | |||
| # sister | |||
| if strings[11] == strings[14]: | |||
| add_format = ":1" | |||
| else: | |||
| add_format = ":0" | |||
| # trioLCL5 | |||
| if strings[15] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # trioLCL6 | |||
| if strings[7] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # filter | |||
| if (strings[11] == strings[14]) and (strings[15] == 'MD=1') and (strings[7] == 'MD=1'): | |||
| strings[6] = 'PASS' | |||
| else: | |||
| strings[6] = '.' | |||
| # output LCL5 | |||
| LCL5outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format + '\n' | |||
| LCL5file.write(LCL5outLine) | |||
| # output LCL6 | |||
| LCL6outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[11] + add_format + '\n' | |||
| LCL6file.write(LCL6outLine) | |||
| # output LCL7 | |||
| LCL7outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[10] + add_format + '\n' | |||
| LCL7file.write(LCL7outLine) | |||
| # output LCL8 | |||
| LCL8outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[9] + add_format + '\n' | |||
| LCL8file.write(LCL8outLine) | |||
| # output family | |||
| familyoutLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+ '.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format +'\t' + strings[11] + add_format + '\t' + strings[10] + add_format +'\t' + strings[9] + add_format + '\n' | |||
| familyfile.write(familyoutLine) | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| process(line) | |||
+ 36
- 0
codescripts/vcf2bed.py
ファイルの表示
| @@ -0,0 +1,36 @@ | |||
| import re | |||
| def position_to_bed(chromo,pos,ref,alt): | |||
| # snv | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF | |||
| if len(ref) == 1 and len(alt) == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| # deletions | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (reference length - alternate length) | |||
| elif len(ref) > 1 and len(alt) == 1: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| #insertions | |||
| # For insertions: | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (alternate length - reference length) | |||
| else: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(alt) - 1) | |||
| return chromo,StartPos,EndPos | |||
| def padding_region(chromo,pos1,pos2,padding): | |||
| StartPos1 = pos1 - padding | |||
| EndPos1 = pos1 | |||
| StartPos2 = pos2 | |||
| EndPos2 = pos2 + padding | |||
| return chromo,StartPos1,EndPos1,StartPos2,EndPos2 | |||
+ 1
- 0
family.tsv
ファイルの表示
| @@ -0,0 +1 @@ | |||
| #LCL5vcf LCL6vcf LCL7vcf LCL8vcf LCL5name LCL6name LCL7name LCL8name familyname | |||
+ 43
- 0
inputs
ファイルの表示
| @@ -0,0 +1,43 @@ | |||
| { | |||
| "{{ project_name }}.LCL7merge.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||
| "{{ project_name }}.LCL6bed_annotation.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL7bed_annotation.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL8bed_annotation.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL7votes.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL6votes.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL5VCFrename.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL8mergeInfo.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL6mendelian.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/vbt:v1.1", | |||
| "{{ project_name }}.LCL5mergeInfo.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.mergeSister.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL5mendelian.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/vbt:v1.1", | |||
| "{{ project_name }}.LCL6allInfozipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL5allInfozipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.disk_size": "150", | |||
| "{{ project_name }}.LCL7mergeInfo.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.inputSamplesFile": "{{ inputSamplesFile }}", | |||
| "{{ project_name }}.repeat_bed": "oss://pgx-result/renluyao/manuscript/all.repeat.bed", | |||
| "{{ project_name }}.LCL6merge.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL6variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||
| "{{ project_name }}.LCL6zipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL7allInfozipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL5votes.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL6mergeInfo.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL5bed_annotation.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL6VCFrename.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL5merge.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL8votes.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.reformVCF.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.1", | |||
| "{{ project_name }}.LCL5zipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.cluster_config": "OnDemand bcs.b4.xlarge img-ubuntu-vpc", | |||
| "{{ project_name }}.LCL8allInfozipIndex.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL7variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||
| "{{ project_name }}.LCL8merge.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-tools:latest", | |||
| "{{ project_name }}.LCL5variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||
| "{{ project_name }}.LCL8variantsNorm.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bcftools:v1.9", | |||
| "{{ project_name }}.ref_dir": "oss://chinese-quartet/quartet-storage-data/reference_data/" | |||
| } | |||
バイナリ
pictures/.DS_Store
ファイルの表示
バイナリ
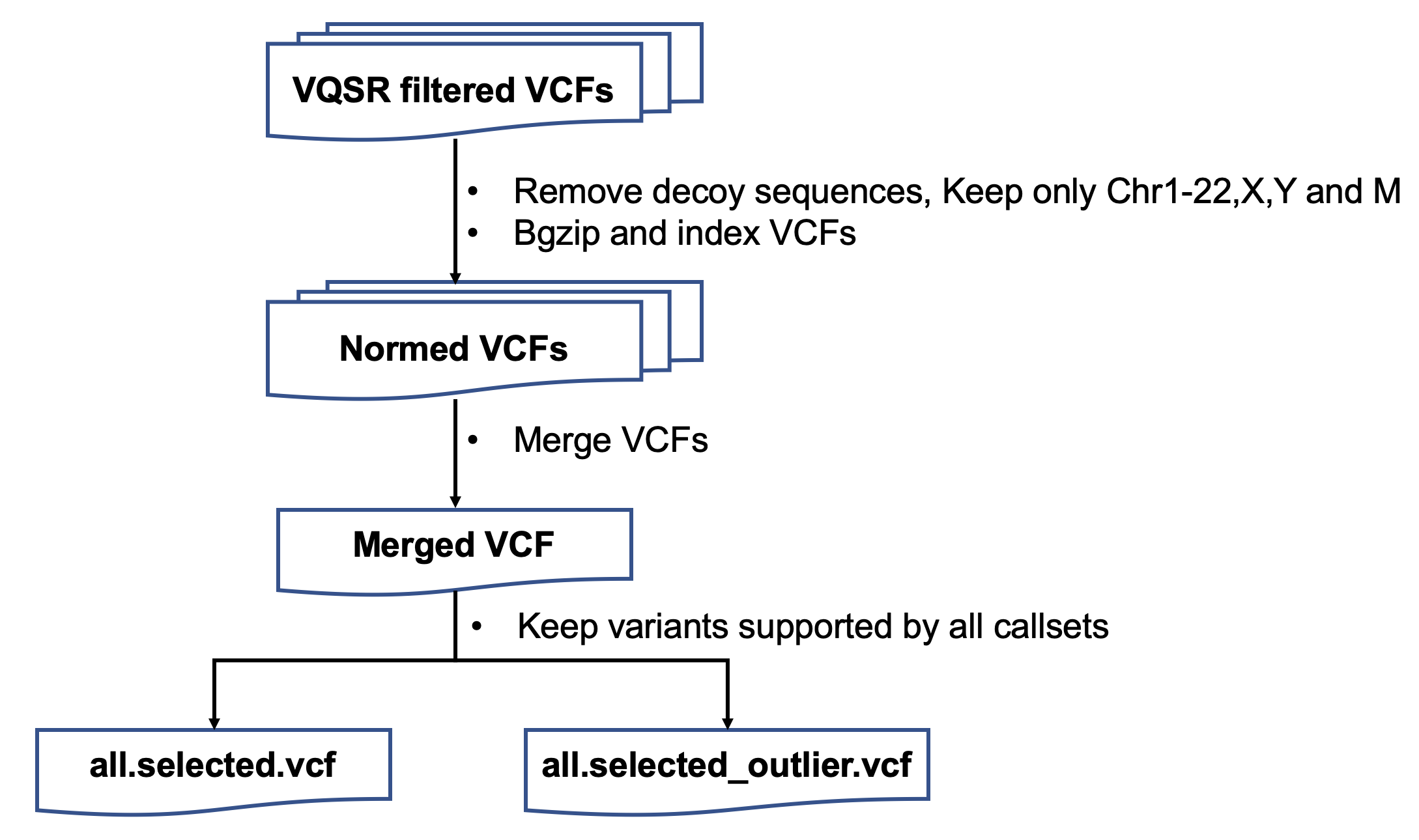
pictures/workflow.png
ファイルの表示
{kind=link}
| 変更前 | 変更後 |
|---|---|
|
|
| 幅: 2178 | 高さ: 1290 | サイズ: 180KB |
バイナリ
tasks/.DS_Store
ファイルの表示
+ 32
- 0
tasks/VCFinfo.wdl
ファイルの表示
| @@ -0,0 +1,32 @@ | |||
| task VCFinfo { | |||
| File repeat_annotated_vcf | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/variants_quality_location_intergration.py -vcf ${repeat_annotated_vcf} -prefix ${sample} | |||
| cat ${sample}_variant_quality_location.txt | grep '#CHROM' > header | |||
| for i in chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr20 chr21 chr22 chrX | |||
| do | |||
| cat ${sample}_variant_quality_location.txt | grep -w $i | cat header - > ${sample}.$i.vcfInfo.txt | |||
| done | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File extracted_info = "${sample}_variant_quality_location.txt" | |||
| Array[File] chromo_vcfInfo = glob("*.vcfInfo.txt") | |||
| } | |||
| } | |||
+ 31
- 0
tasks/VCFrename.wdl
ファイルの表示
| @@ -0,0 +1,31 @@ | |||
| task VCFrename { | |||
| File trio_vcf_gz | |||
| File trio_vcf_idx | |||
| String mother_name | |||
| String father_name | |||
| String child_name | |||
| String family_name | |||
| String child | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| echo "MOTHER ${mother_name}.${child} | |||
| FATHER ${father_name}.${child} | |||
| CHILD ${child_name}" > rename.txt | |||
| rtg vcfannotate -i ${trio_vcf_gz} -o ${family_name}.${child}.rename.vcf.gz --relabel=rename.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File rename_trio_vcf_gz = "${family_name}.${child}.rename.vcf.gz" | |||
| File rename_trio_vcf_idx = "${family_name}.${child}.rename.vcf.gz.tbi" | |||
| } | |||
| } | |||
+ 27
- 0
tasks/bed_annotation.wdl
ファイルの表示
| @@ -0,0 +1,27 @@ | |||
| task bed_annotation { | |||
| File merged_vcf_gz | |||
| File merged_vcf_idx | |||
| File repeat_bed | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| rtg vcfannotate --bed-info=${repeat_bed} -i ${merged_vcf_gz} -o ${sample}.mendelian.merged.repeatAnno.vcf.gz | |||
| gunzip ${sample}.mendelian.merged.repeatAnno.vcf.gz | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File repeat_annotated_vcf = "${sample}.mendelian.merged.repeatAnno.vcf" | |||
| } | |||
| } | |||
+ 26
- 0
tasks/final_result.wdl
ファイルの表示
| @@ -0,0 +1,26 @@ | |||
| task FinalResult { | |||
| File extracted_info | |||
| File annotated_txt | |||
| String prefix = basename(annotated_txt,".mendelian.txt") | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/FinalResult2VCF.py -vcfInfo ${extracted_info} -mendelianInfo ${annotated_txt} -prefix ${prefix} -sample ${sample} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File benchmarking_calls = "${prefix}_benchmarking_calls.vcf" | |||
| File all_info = "${prefix}_all_sample_information.vcf" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/mendelian.wdl
ファイルの表示
| @@ -0,0 +1,34 @@ | |||
| task mendelian { | |||
| File child_vcf | |||
| File LCL7_vcf | |||
| File LCL8_vcf | |||
| String LCL7_name | |||
| String LCL8_name | |||
| String child_name | |||
| File ref_dir | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||
| nt=$(nproc) | |||
| mkdir VBT | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${LCL8_vcf} -father ${LCL7_vcf} -child ${child_vcf} -outDir VBT -out-prefix ${child_name}.family --output-violation-regions -thread-count $nt | |||
| cat VBT/${child_name}.family_trio.vcf > ${child_name}.family.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| Array[File] vbt_mendelian = glob("VBT/*") | |||
| File trio_vcf = "${child_name}.family.vcf" | |||
| } | |||
| } | |||
+ 28
- 0
tasks/merge.wdl
ファイルの表示
| @@ -0,0 +1,28 @@ | |||
| task merge { | |||
| Array[File] family_vcf_gz | |||
| Array[File] family_vcf_idx | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| rtg vcfmerge --force-merge-all -o ${sample}.merged.vcf.gz ${sep=" " family_vcf_gz} | |||
| zcat ${sample}.merged.vcf.gz | grep -v '#' | cut -f1-2 | sed s'/\t/_/g' | sort | uniq -c | sed 's/\s\+/\t/g' | awk '{ if ($1 != 1) { print } }' | cut -f3 > ${sample}.vcf_dup.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File merged_vcf_gz = "${sample}.merged.vcf.gz" | |||
| File merged_vcf_idx = "${sample}.merged.vcf.gz.tbi" | |||
| File vcf_dup = "${sample}.vcf_dup.txt" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/mergeSister.wdl
ファイルの表示
| @@ -0,0 +1,34 @@ | |||
| task mergeSister { | |||
| File LCL5_trio_vcf_gz | |||
| File LCL5_trio_vcf_idx | |||
| File LCL6_trio_vcf_gz | |||
| File LCL6_trio_vcf_idx | |||
| String family_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| rtg vcfmerge -o LCL5.LCL6.merged.vcf.gz ${LCL5_trio_vcf_gz} ${LCL6_trio_vcf_gz} | |||
| rtg vcfmerge -o LCL6.LCL5.merged.vcf.gz ${LCL6_trio_vcf_gz} ${LCL5_trio_vcf_gz} | |||
| zcat LCL5.LCL6.merged.vcf.gz | grep '##' > header | |||
| zcat LCL5.LCL6.merged.vcf.gz | grep -v '##' | cut -f8 > LCL5.mendelian | |||
| zcat LCL6.LCL5.merged.vcf.gz | grep -v '##' | paste - LCL5.mendelian > body | |||
| cat header body > ${family_name}.trio.info.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File family_mendelian_info = "${family_name}.trio.info.vcf" | |||
| } | |||
| } | |||
+ 25
- 0
tasks/mergeVCFInfo.wdl
ファイルの表示
| @@ -0,0 +1,25 @@ | |||
| task mergeVCFInfo { | |||
| Array[File] vcf_gz | |||
| Array[File] vcf_idx | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| rtg vcfmerge --force-merge-all -o ${sample}.merged.info.vcf.gz ${sep=" " vcf_gz} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File merged_vcf = "${sample}.merged.info.vcf.gz" | |||
| File merged_vcf_idx = "${sample}.merged.info.vcf.gz.tbi" | |||
| } | |||
| } | |||
+ 24
- 0
tasks/merge_info.wdl
ファイルの表示
| @@ -0,0 +1,24 @@ | |||
| task merge_info { | |||
| File vcfInfo | |||
| File mendelianInfo | |||
| String sample | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/merge_mendelian_vcfinfo.py -vcfInfo ${vcfInfo} -mendelianInfo ${mendelianInfo} -sample ${sample} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File all_info = "${sample}_mendelian_vcfInfo.vcf" | |||
| } | |||
| } | |||
+ 39
- 0
tasks/oneClass.wdl
ファイルの表示
| @@ -0,0 +1,39 @@ | |||
| task oneClass { | |||
| File snv_train_vcf | |||
| File snv_test_vcf | |||
| File indel_train_vcf | |||
| File indel_test_vcf | |||
| String sampleName = basename(snv_train_vcf,".normed.snv.train.txt") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/oneClass.py -train ${snv_train_vcf} -test ${snv_test_vcf} -name ${sampleName}_snv | |||
| python /opt/oneClass.py -train ${indel_train_vcf} -test ${indel_test_vcf} -name ${sampleName}_indel | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File snv_true_txt = "${sampleName}_snv_predicted_true.txt" | |||
| File snv_false_txt = "${sampleName}_snv_predicted_false.txt" | |||
| File snv_true_bed = "${sampleName}_snv_predicted_true.bed" | |||
| File snv_false_bed = "${sampleName}_snv_predicted_false.bed" | |||
| File snv_padding = "${sampleName}_snv_padding.bed" | |||
| File indel_true_txt = "${sampleName}_indel_predicted_true.txt" | |||
| File indel_false_txt = "${sampleName}_indel_predicted_false.txt" | |||
| File indel_true_bed = "${sampleName}_indel_predicted_true.bed" | |||
| File indel_false_bed = "${sampleName}_indel_predicted_false.bed" | |||
| File indel_padding = "${sampleName}_indel_padding.bed" | |||
| } | |||
| } | |||
+ 38
- 0
tasks/reformVCF.wdl
ファイルの表示
| @@ -0,0 +1,38 @@ | |||
| task reformVCF { | |||
| File family_mendelian_info | |||
| File family_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/reformVCF.py -vcf ${family_mendelian_info} -name ${family_name} | |||
| cat ${family_name}.LCL5.vcf | grep -v '##' | grep -v '0/0' > ${family_name}.LCL5.txt | |||
| cat ${family_name}.LCL6.vcf | grep -v '##' | grep -v '0/0' > ${family_name}.LCL6.txt | |||
| cat ${family_name}.LCL7.vcf | grep -v '##' | grep -v '0/0' > ${family_name}.LCL7.txt | |||
| cat ${family_name}.LCL8.vcf | grep -v '##' | grep -v '0/0' > ${family_name}.LCL8.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File LCL5_family_info = "${family_name}.LCL5.vcf" | |||
| File LCL6_family_info = "${family_name}.LCL6.vcf" | |||
| File LCL7_family_info = "${family_name}.LCL7.vcf" | |||
| File LCL8_family_info = "${family_name}.LCL8.vcf" | |||
| File family_info = "${family_name}.vcf" | |||
| File LCL5_family_info_txt = "${family_name}.LCL5.txt" | |||
| File LCL6_family_info_txt = "${family_name}.LCL6.txt" | |||
| File LCL7_family_info_txt = "${family_name}.LCL7.txt" | |||
| File LCL8_family_info_txt = "${family_name}.LCL8.txt" | |||
| } | |||
| } | |||
+ 35
- 0
tasks/sister.wdl
ファイルの表示
| @@ -0,0 +1,35 @@ | |||
| task sister { | |||
| File LCL5_vcf | |||
| File LCL6_vcf | |||
| File ref_dir | |||
| String fasta | |||
| String family_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||
| mkdir sister | |||
| /opt/VBT-TrioAnalysis/vbt varcomp -called ${LCL5_vcf} -base ${LCL6_vcf} -ref ${ref_dir}/${fasta} -outDir sister -filter none | |||
| mv sister/TPBase.vcf ${family_name}.sister.consistent.vcf | |||
| mv sister/FP.vcf ${family_name}.LCL5.uniq.vcf | |||
| mv sister/FN.vcf ${family_name}.LCL6.uniq.vcf | |||
| mv sister/log.txt ${family_name}.sister.vbt.log.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File sister_consistent_vcf = "${family_name}.sister.consistent.vcf" | |||
| File LCL5_uniq = "${family_name}.LCL5.uniq.vcf" | |||
| File LCL6_uniq = "${family_name}.LCL6.uniq.vcf" | |||
| File log = "${family_name}.sister.vbt.log.txt" | |||
| } | |||
| } | |||
+ 33
- 0
tasks/variantsNorm.wdl
ファイルの表示
| @@ -0,0 +1,33 @@ | |||
| task variantsNorm { | |||
| File vcf | |||
| File ref_dir | |||
| String fasta | |||
| String sampleName | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${vcf} | grep '#' > header | |||
| cat ${vcf} | grep -v '#' > body | |||
| cat body | grep -w '^chr1\|^chr2\|^chr3\|^chr4\|^chr5\|^chr6\|^chr7\|^chr8\|^chr9\|^chr10\|^chr11\|^chr12\|^chr13\|^chr14\|^chr15\|^chr16\|^chr17\|^chr18\|^chr19\|^chr20\|^chr21\|^chr22\|^chrX' > body.filtered | |||
| cat header body.filtered > ${sampleName}.filtered.vcf | |||
| /opt/hall-lab/bcftools-1.9/bin/bcftools norm -f ${ref_dir}/${fasta} ${sampleName}.filtered.vcf > ${sampleName}.normed.vcf | |||
| cat ${sampleName}.normed.vcf | grep -v '##' > ${sampleName}.normed.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File normed_vcf = "${sampleName}.normed.vcf" | |||
| File normed_txt = "${sampleName}.normed.txt" | |||
| } | |||
| } | |||
+ 26
- 0
tasks/votes.wdl
ファイルの表示
| @@ -0,0 +1,26 @@ | |||
| task votes { | |||
| File repeat_annotated_vcf | |||
| File vcf_dup | |||
| String sample | |||
| String prefix | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/high_confidence_call_vote.py -vcf ${repeat_annotated_vcf} -dup ${vcf_dup} -sample ${sample} -prefix ${prefix} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File voted_vcf = "${prefix}_voted.vcf" | |||
| File all_sample_vcf = "${prefix}_all_sample_information.vcf" | |||
| } | |||
| } | |||
+ 24
- 0
tasks/zipIndex.wdl
ファイルの表示
| @@ -0,0 +1,24 @@ | |||
| task zipIndex { | |||
| File vcf | |||
| String vcf_name = basename(vcf,".vcf") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| rtg bgzip ${vcf} -c > ${vcf_name}.vcf.gz | |||
| rtg index -f vcf ${vcf_name}.vcf.gz | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf_gz = "${vcf_name}.vcf.gz" | |||
| File vcf_idx = "${vcf_name}.vcf.gz.tbi" | |||
| } | |||
| } | |||
+ 92
- 0
workflow.wdl
ファイルの表示
| @@ -0,0 +1,92 @@ | |||
| import "./tasks/mendelian.wdl" as mendelian | |||
| workflow {{ project_name }} { | |||
| File inputSamplesFile | |||
| Array[Array[File]] inputSamples = read_tsv(inputSamplesFile) | |||
| File ref_dir | |||
| String fasta | |||
| String cluster_config | |||
| String disk_size | |||
| scatter (quartet in inputSamples){ | |||
| call mendelian.mendelian as LCL5mendelian { | |||
| input: | |||
| child_vcf=quartet[0], | |||
| LCL7_vcf=quartet[2], | |||
| LCL8_vcf=quartet[3], | |||
| LCL7_name=quartet[6], | |||
| LCL8_name=quartet[7], | |||
| child_name=quartet[4], | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mendelian.mendelian as LCL6mendelian { | |||
| input: | |||
| child_vcf=quartet[1], | |||
| LCL7_vcf=quartet[2], | |||
| LCL8_vcf=quartet[3], | |||
| LCL7_name=quartet[6], | |||
| LCL8_name=quartet[7], | |||
| child_name=quartet[5], | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call zipIndex.zipIndex as LCL5zipIndex { | |||
| input: | |||
| vcf=LCL5mendelian.trio_vcf, | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call zipIndex.zipIndex as LCL6zipIndex { | |||
| input: | |||
| vcf=LCL6mendelian.trio_vcf, | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call VCFrename.VCFrename as LCL5VCFrename { | |||
| input: | |||
| trio_vcf_gz=LCL5zipIndex.vcf_gz, | |||
| trio_vcf_idx=LCL5zipIndex.vcf_idx, | |||
| mother_name=quartet[7], | |||
| father_name=quartet[6], | |||
| child_name=quartet[4], | |||
| family_name=quartet[8], | |||
| child="LCL5", | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call VCFrename.VCFrename as LCL6VCFrename { | |||
| input: | |||
| trio_vcf_gz=LCL6zipIndex.vcf_gz, | |||
| trio_vcf_idx=LCL6zipIndex.vcf_idx, | |||
| mother_name=quartet[7], | |||
| father_name=quartet[6], | |||
| child_name=quartet[5], | |||
| family_name=quartet[8], | |||
| child="LCL6", | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mergeSister.mergeSister as mergeSister { | |||
| input: | |||
| LCL5_trio_vcf_gz=LCL5VCFrename.rename_trio_vcf_gz, | |||
| LCL5_trio_vcf_idx=LCL5VCFrename.rename_trio_vcf_idx, | |||
| LCL6_trio_vcf_gz=LCL6VCFrename.rename_trio_vcf_gz, | |||
| LCL6_trio_vcf_idx=LCL6VCFrename.rename_trio_vcf_idx, | |||
| family_name=quartet[8], | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call reformVCF.reformVCF as reformVCF { | |||
| input: | |||
| family_mendelian_info=mergeSister.family_mendelian_info, | |||
| family_name=quartet[8], | |||
| cluster_config=cluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| } | |||
読み込み中…