#中华家系1号标准物质室间质评报告系统分析流程
> Author: Run Luyao
>
> E-mail:18110700050@fudan.edu.cn
>
> Git: http://choppy.3steps.cn/renluyao/Quality_control.git
>
> Last Updates: 30/8/2019
## 安装指南
```
# 激活choppy环境
source activate choppy
# 安装app
choppy install renluyao/Quality_control
```
## App概述——中华家系1号标准物质介绍
建立高通量全基因组测序的生物计量和质量控制关键技术体系,是保障测序数据跨技术平台、跨实验室可比较、相关研究结果可重复、数据可共享的重要关键共性技术。建立国家基因组标准物质和基准数据集,突破基因组学的生物计量技术,是将测序技术转化成临床应用的重要环节与必经之路,目前国际上尚属空白。中国计量科学研究院与复旦大学、复旦大学泰州健康科学研究院共同研制了人源中华家系1号基因组标准物质(**Quartet,一套4个样本,编号分别为LCL5,LCL6,LCL7,LCL8,其中LCL5和LCL6为同卵双胞胎女儿,LCL7为父亲,LCL8为母亲**),以及相应的全基因组测序序列基准数据集(“量值”),为衡量基因序列检测准确与否提供一把“标尺”,成为保障基因测序数据可靠性的国家基准。人源中华家系1号基因组标准物质来源于泰州队列同卵双生双胞胎家庭,从遗传结构上体现了我国南北交界的人群结构特征,同时家系的设计也为“量值”的确定提供了遗传学依据。
中华家系1号DNA标准物质的标称值包括高置信单核苷酸变异信息、高置信短插入缺失变异信息和77.9-78.1%的高置信参考基因组区。该系列标准物质可以用于评估基因组测序的性能,包括全基因组测序、全外显子测序、靶向测序,如基因捕获测序;还可用于评估测序过程和数据分析过程中对SNV和InDel检出的真阳性、假阳性、真阴性和假阴性水平,为基因组测序技术平台、实验室、相关产品的质量控制与性能验证提供标准物质和标准数据。
该Quality_control APP用于全基因组测序(whole-genome sequencing,WGS)数据的质量评估,包括原始数据质控、比对数据质控和突变检出数据质控。
## 流程与参数
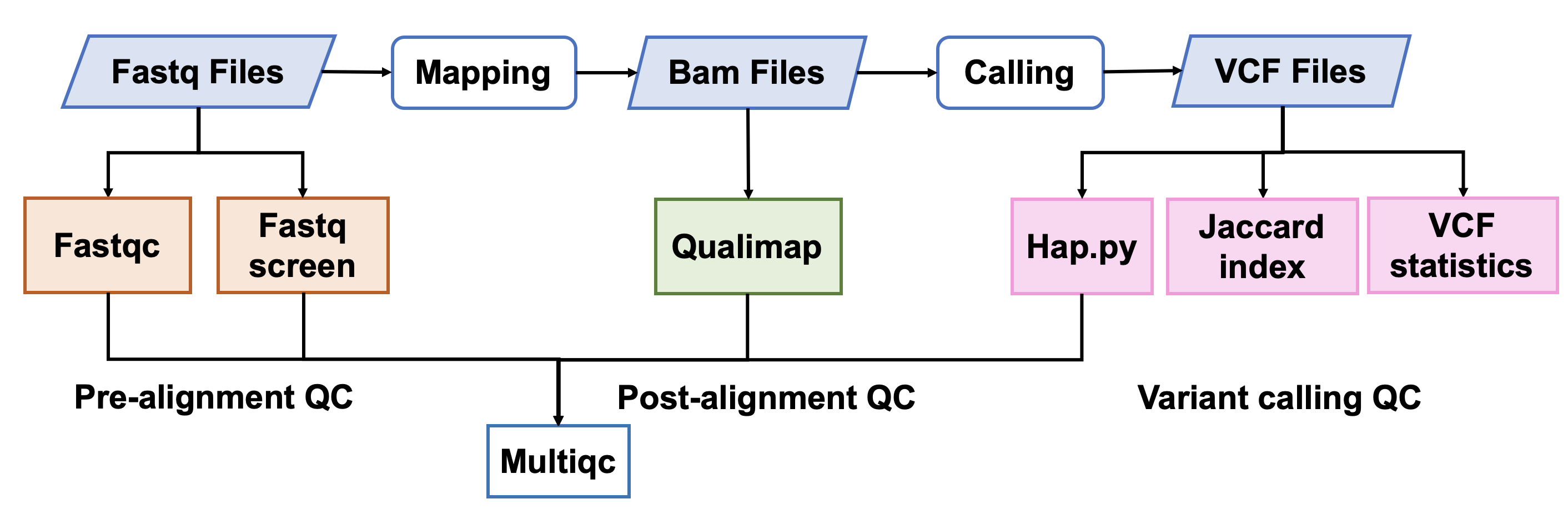
###1. 原始数据质量控制
#### [Fastqc]() v0.11.5
FastQC是一个常用的测序原始数据的质控软件,主要包括12个模块,具体请参考[Fastqc模块详情]()。
```bash
fastqc -t -o
```
#### [Fastq Screen]() 0.12.0
Fastq Screen是检测测序原始数据中是否引⼊入其他物种,或是接头引物等污染,⽐比如,如果测序样本
是⼈人类,我们期望99%以上的reads匹配到⼈人类基因组,10%左右的reads匹配到与⼈人类基因组同源性
较⾼高的⼩小⿏鼠上。如果有过多的reads匹配到Ecoli或者Yeast,要考虑是否在培养细胞的时候细胞系被污
染,或者建库时⽂文库被污染。
````bash
fastq_screen --aligner --conf --top --threads
````
`--conf` conifg 文件主要输入了多个物种的fasta文件地址,可根据自己自己的需求下载其他物种的fasta文件加入分析
`--top`一般不需要对整个fastq文件进行检索,取前100000行
###2. 比对后数据质量控制
#### [Qualimap]() 2.0.0
Qualimap是一个比对指控软件,包含Picard的MarkDuplicates的结果和sentieon中metrics的质控结果。
```bash
qualimap bamqc -bam -outformat PDF:HTML -nt -outdir --java-mem-size=32G
```
###3. 突变检出数据质量控制
#### [Hap.py]() v0.3.9
hap.py是将被检测vcf结果与benchmarking对比,计算precision和recall的软件,它考虑了vcf中[突变表示形式的多样性](),进行了归一化。
```bash
hap.py -f --threads -o
```
#### [Jaccard index]() (rtg-tools 3.10.1)
Jaccard index是两个集合的交集除以两个集合的并集。这个计算用的是rtg-tools的vcfeval,它将两个vcf中的突变表示格式进行了统一,即两个看似不同的SNV或者INDEL实际上是指同一个突变,这种情况经常发生在重复区域。
```bash
rtg vcfeval -b -c -o -t
```
`-t` sdf文件是rtg-tools专用的注释文件,由fasta文件转换来
#### [VCF statistics]() (rtg-tools 3.10.1)
该计算用的是rtg-tools的vcfstats,对vcf中的SNV和INDEL的数目进行了统计。
```bash
rtg vcfstats
```
### 4. 质控数据整合
####[MultiQC]() v1.8
以上的质控软件的输出都是单个文件的质控结果,multiqc可以将这些结果进行汇总,以及网页可视化展示
```bash
multiqc
```
## App输入变量与输入文件
在安装了APP之后,输入一下命令得到需要准备的文件
```bash
choppy samples Quality_control-latest --output qcsamples
```
qcsamples文件
```bash
inputJIpiarsFile,inputSamplesFile,sample_id
oss://pgx-result/renluyao/inputJIpiarsFileExample.tsv,oss://pgx-result/renluyao/inputSamplesFileExamples.tsv,1
```
**inputJIpiarsFile**是计算Jaccard index的输入文件,按以下格式进行准备
```bash
#vcf1 #vcf2 #vcf1_vcf2
oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/72f269f2-91b7-4fbe-bde7-99b2e1e3091c/call-Haplotyper/Fudan_DNA_LCL7_hc.vcf oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Haplotyper/Fudan_DNA_LCL5_hc.vcf LCL7_LCL5
```
`vcf1`和` vcf2`是两个需要计算一致性的vcf文件的oss地址
`vcf1_vcf2`是两个vcf文件名字的简单缩写,用于之后的数据分析
**inputSamplesFile**是其他质控task的输入文件,按以下格式进行准备
```bash
#fastq_read1 #fastq_read2 #bam #bai #vcf #sample_mark
oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R1.fastq.gz oss://chinese-quartet/quartet-test-data/fastqfiles/Fudan_DNA_LCL5_R2.fastq.gz oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Dedup/Fudan_DNA_LCL5.sorted.deduped.bam.bai oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Haplotyper/Fudan_DNA_LCL5_hc.vcf LCL5
```
`fastq_read1`和`fastq_read2`是两个fastq文件的oss地址
`bam`是bam文件的oss地址
`bai`是bam文件的索引文件的oss地址
`vcf`是vcf文件的地址
`sample_mark`是标识测序数据的样本,可填写LCL5、LCL6、LCL7和LCL8
**sample_id**是choppy app内置的识别标志,写1即可
## App输出文件
以下task的输出都只包含了单个文件的质控结果
**fastq.wdl**
- _fastqc.html
- _fastqc.zip
**fastqscreen.wdl**
- _screen.png
- _screen.txt
- _screen.html
**bamqc.wdl**
- _qualimap.zip
**benchmark.wdl**
- .rtg.vcf.gz
- .rtg.vcf.gz.tbi
- .vcf.gz
- .vcf.gz.tbi
- .roc.all.csv.gz
- .roc.Locations.INDEL.csv.gz
- .roc.Locations.INDEL.PASS.csv.gz
- .roc.Locations.SNP.csv.gz
- .roc.Locations.SNP.PASS.csv.gz
- .summary.csv
- .extended.csv
- .metrics.json.gz
**jaccard_index.wdl**
- summary.txt
**vcfstat.wdl**
- onestats.txt
主要查看以下三个task的结果,汇总了所有样本的结果
**mergeJI.wdl**
- result.txt
**mergeNum.wdl**
- vcfstats.txt
**multiqc**
- multiqc_report.html
- multiqc.log
- multiqc_data.json
- multiqc_fastq_screen.txt
- multiqc_fastqc.txt
- multiqc_general_stats.txt
- multiqc_happy_data.json
- multiqc_sources.txt
## 结果展示与解读
#### 1. result.txt
```bash
#vcf1 #vcf2 #vcf1_vcf2 #True-pos-call-number #False-pos-number #False-neg-number
oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/72f269f2-91b7-4fbe-bde7-99b2e1e3091c/call-Haplotyper/Fudan_DNA_LCL7_hc.vcf oss://choppy-cromwell-result/test-choppy/wgs_quartettest_renluyao_0827/7a72d0e6-302d-43ca-b6b0-daeaa0236d06/call-Haplotyper/Fudan_DNA_LCL5_hc.vcf LCL7_LCL5 4891550 11116 11116
```
在获得result.txt之后,jaccard index的计算公式如下
$$
JaccardIndex=\frac{TP}{TP+FP+FN}
$$
#### 2. vcfstats.txt
```bash
File Failed Filters Passed Filters SNPs MNPs Insertions Deletions Indels Same as reference SNP Transitions/Transversions Total Het/Hom ratio SNP Het/Hom ratio MNP Het/Hom ratio Insertion Het/Hom ratio Deletion Het/Hom ratio Indel Het/Hom ratio Insertion/Deletion ratio Indel/SNP+MNP ratio
/cromwell_inputs/choppy-cromwell-result/test-choppy/qc_test_renluyao_0831/79830609-9bd2-4e0c-9483-0c3369052a9d/call-benchmark/shard-0/Fudan_DNA_LCL5_hc.rtg.vcf.gz 0 4904087 4024591 0 421154 445839 12503 0 1.98 (3771780/1907484) 1.44 (2897278/2006809) 1.43 (2371579/1653012) - (0/0) 1.37 (243816/177338) 1.53 (269380/176459) - (12503/0) 0.94 (421154/445839) 0.22 (879496/4024591)
```
#### 3. MultiQC输出的结果
下载之后将文件名glob**改成multiqc_data,即可打开multiqc_report.html查看可视化的结果,在multiqc_data中的整合结果txt文件,可以用于报告系统的输入。
对应的fastqc、fastqscreen、qualimap、hap.py的结果解释请查询对应的官网。
## CHANGELOG
**Version 1.0 - Auguest 30, 2019**
- 完成PGx常规质控流程的choppy APP
## FAQ
**1. RNAseq和甲基化的质控流程?**
可查询multiqc支持的质控模块
RNAseq和甲基化的质控流程待完善
**2. 如果样本没有技术重复,该APP中的inputJIpiarsFile是怎么输入的?**
在Version 1.0中暂时还没有考虑没有技术重复的问题,可输入姐妹、父母、父女、母女的配对,计算同卵双胞胎、亲属关系和陌生人之间基因突变位点的一致性。
**3. 怎么对该APP的输出结果进行可视化?**
正在努力开发中
**4. bam文件和vcf文件怎么获得?**
在进行质控分析前,请先用标准化流程进行测序数据分析,详情查看choppy APP [huangyechao/wgs-germline]()