
 LUYAO REN
пре 2 година
LUYAO REN
пре 2 година
комит
ad73c9d27a
60 измењених фајлова са 5029 додато и 0 уклоњено
+ 251
- 0
README.md
Прегледај датотеку
| @@ -0,0 +1,251 @@ | |||
| # Quality control of germline variants calling results using a Chinese Quartet family | |||
| > Author: Run Luyao | |||
| > | |||
| > E-mail:18110700050@fudan.edu.cn | |||
| > | |||
| > Git: http://47.103.223.233/renluyao/quartet_dna_quality_control_wgs_big_pipeline | |||
| > | |||
| > Last Updates: 2022/10/31 | |||
| ## Install | |||
| ``` | |||
| open-choppy-env | |||
| choppy install renluyao/quartet_dna_quality_control_big_pipeline | |||
| ``` | |||
| ## Introduction | |||
| ###Chinese Quartet DNA reference materials | |||
| With the rapid development of sequencing technology and the dramatic decrease of sequencing costs, DNA sequencing has been widely used in scientific research, diagnosis of and treatment selection for human diseases. However, due to the lack of effective quality assessment and control of the high-throughput omics data generation and analysis processes, variants calling results are seriously inconsistent among different technical replicates, batches, laboratories, sequencing platforms, and analysis pipelines, resulting in irreproducible scientific results and conclusions, huge waste of resources, and even endangering the life and health of patients. Therefore, reference materials for quality control of the whole process from omics data generation to data analysis are urgently needed. | |||
| We first established genomic DNA reference materials from four immortalized B-lymphoblastoid cell lines of a Chinese Quartet family including parents and monozygotic twin daughters to make performance assessment of germline variants calling results. To establish small variant benchmark calls and regions, we generated whole-genome sequencing data in nine batches, with depth ranging from 30x to 60x, by employing PCR-free and PCR libraries on four popular short-read sequencing platforms (Illumina HiSeq XTen, Illumina NovaSeq, MGISEQ-2000, and DNBSEQ-T7) with three replicates at each batch, resulting in 108 libraries in total and 27 libraries for each Quartet DNA reference material. Then, we selected variants concordant in multiple call sets and in Mendelian consistency within Quartet family members as small variant benchmark calls, resulting in 4.2 million high-confidence variants (SNV and Indel) and 2.66 G high confidence genomic region, covering 87.8% of the human reference genome (GRCh38, chr1-22 and X). Two orthogonal technologies were used for verifying the high-confidence variants. The consistency rate with PMRA (Axiom Precision Medicine Research Array) was 99.6%, and 95.9% of high-confidence variants were validated by 10X Genomics whole-genome sequencing data. Genetic built-in truth of the Quartet family design is another kind of “truth” within the four Quartet samples. Apart from comparison with benchmark calls in the benchmark regions to identify false-positive and false-negative variants, pedigree information among the Quartet DNA reference materials, i.e., reproducibility rate of variants between the twins and Mendelian concordance rate among family members, are complementary approaches to comprehensively estimate genome-wide variants calling performance. Finally, we developed a whole-genome sequencing data quality assessment pipeline and demonstrated its utilities with two examples of using the Quartet reference materials and datasets to evaluate data generation performance in three sequencing labs and different data analysis pipelines. | |||
| ### Quality control pipeline for WGS | |||
| This Quartet quality control pipeline evaluate the performance of reads quality and variant calling quality. This pipeline accepts FASTQ format input files or VCF format input files. If the users input FASTQ files, this APP will output the results of pre-alignment quality control from FASTQ files, post-alignment quality control from BAM files and variant calling quality control from VCF files. [GATK best practice pipelines](https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels-) (implemented by [SENTIEON software](https://support.sentieon.com/manual/)) were used to map reads to the reference genome and call variants. If the users input VCF files, this APP will only output the results of variant calling quality control. | |||
| Quartet quality control analysis pipeline started from FASTQ files is implemented across seven main procedures: | |||
| - Pre-alignment QC of FASTQ files | |||
| - Genome alignment | |||
| - Post-alignment QC of BAM files | |||
| - Germline variant calling | |||
| - Variant calling QC depended on benchmark sets of VCF files | |||
| - Check Mendelian ingeritance states across four Quartet samples of every variants | |||
| - Variant calling QC depended on Quartet genetic relationship of VCF files | |||
| Quartet quality control analysis pipeline started from VCF files is implemented across three main procedures: | |||
| - Variant calling QC depended on benchmark sets of VCF files | |||
| - Check Mendelian ingeritance states across four Quartet samples of every variants | |||
| - Variant calling QC depended on Quartet genetic relationship of VCF files | |||
|  | |||
| Results generated from this APP can be visualized by Choppy report. | |||
| ## Data Processing Steps | |||
| ### 1. Pre-alignment QC of FASTQ files | |||
| #### [Fastqc](<https://www.bioinformatics.babraham.ac.uk/projects/fastqc/>) v0.11.5 | |||
| [FastQC](<https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3%20Analysis%20Modules/>) is used to investigate the quality of fastq files | |||
| ```bash | |||
| fastqc -t <threads> -o <output_directory> <fastq_file> | |||
| ``` | |||
| #### [Fastq Screen](<https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/>) 0.12.0 | |||
| Fastq Screen is used to inspect whether the library were contaminated. For example, we expected 99% reads aligned to human genome, 10% reads aligned to mouse genome, which is partly homologous to human genome. If too many reads are aligned to E.Coli or Yeast, libraries or cell lines are probably comtminated. | |||
| ```bash | |||
| fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file> | |||
| ``` | |||
| ### 2. Genome alignment | |||
| ####[sentieon-genomics](https://support.sentieon.com/manual/):v2019.11.28 | |||
| Reads were mapped to the human reference genome GRCh38 using Sentieon BWA. | |||
| ```bash | |||
| ${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i - | |||
| ``` | |||
| ### 3. Post-alignment QC | |||
| Qualimap and Paicard Tools (implemented by Sentieon) are used to check the quality of BAM files. Deduplicated BAM files are used in this step. | |||
| #### [Qualimap](<http://qualimap.bioinfo.cipf.es/>) 2.0.0 | |||
| ```bash | |||
| qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G | |||
| ``` | |||
| ####[Sentieon-genomics](https://support.sentieon.com/manual/):v2019.11.28 | |||
| ``` | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt --algo QualityYield ${sample}_deduped_QualityYield.txt --algo WgsMetricsAlgo ${sample}_deduped_WgsMetricsAlgo.txt | |||
| ``` | |||
| ### 4. Germline variant calling | |||
| HaplotyperCaller implemented by Sentieon is used to identify germline variants. | |||
| ```bash | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper ${sample}_hc.vcf | |||
| ``` | |||
| ### 5. Variants Calling QC | |||
|  | |||
| #### 5.1 Performance assessment based on benchmark sets | |||
| #### [Hap.py](<https://github.com/Illumina/hap.py>) v0.3.9 | |||
| Variants were compared with benchmark calls in benchmark regions. | |||
| ```bash | |||
| hap.py <truth_vcf> <query_vcf> -f <bed_file> --threads <threads> -o <output_filename> | |||
| ``` | |||
| #### 5.2 Performance assessment based on Quartet genetic built-in truth | |||
| #### [VBT](https://github.com/sbg/VBT-TrioAnalysis) v1.1 | |||
| We splited the Quartet family to two trios (F7, M8, D5 and F7, M8, D6) and then do the Mendelian analysis. A Quartet Mendelian concordant variant is the same between the twins (D5 and D6) , and follow the Mendelian concordant between parents (F7 and M8). Mendelian concordance rate is the Mendelian concordance variant divided by total detected variants in a Quartet family. Only variants on chr1-22,X are included in this analysis. | |||
| ```bash | |||
| vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads> | |||
| ``` | |||
| ## Input files | |||
| ```bash | |||
| choppy samples renluyao/quartet_dna_quality_control_wgs_big_pipeline-latest --output samples | |||
| ``` | |||
| ####Samples CSV file | |||
| #### 1. Start from Fastq files | |||
| ```BASH | |||
| sample_id,project,fastq_1_D5,fastq_2_D5,fastq_1_D6,fastq_2_D6,fastq_1_F7,fastq_2_F7,fastq_1_M8,fastq_2_M8 | |||
| # sample_id in choppy system | |||
| # project name | |||
| # oss path of D5 fastq read1 file | |||
| # oss path of D5 fastq read2 file | |||
| # oss path of D6 fastq read1 file | |||
| # oss path of D6 fastq read2 file | |||
| # oss path of F7 fastq read1 file | |||
| # oss path of F7 fastq read2 file | |||
| # oss path of M8 fastq read1 file | |||
| # oss path of M8 fastq read2 file | |||
| ``` | |||
| #### 2. Start from VCF files | |||
| ```BASH | |||
| sample_id,project,vcf_D5,vcf_D6,vcf_F7,vcf_M8 | |||
| # sample_id in choppy system | |||
| # project name | |||
| # oss path of D5 VCF file | |||
| # oss path of D6 VCF file | |||
| # oss path of F7 VCF file | |||
| # oss path of M8 VCF file | |||
| ``` | |||
| ## Output Files | |||
| #### 1. extract_tables.wdl/extract_tables_vcf.wdl | |||
| (FASTQ) Pre-alignment QC: pre_alignment.txt | |||
| (FASTQ) Post-alignment QC: post_alignment.txt | |||
| (FASTQ/VCF) Variants calling QC: variants.calling.qc.txt | |||
| ####2. quartet_mendelian.wdl | |||
| (FASTQ/VCF) Variants calling QC: mendelian.txt | |||
| ## Output files format | |||
| ####1. pre_alignment.txt | |||
| | Column name | Description | | |||
| | ------------------------- | --------------------------------------------------------- | | |||
| | Sample | Sample name | | |||
| | %Dup | Percentage duplicate reads | | |||
| | %GC | Average GC percentage | | |||
| | Total Sequences (million) | Total sequences | | |||
| | %Human | Percentage of reads mapped to human genome | | |||
| | %EColi | Percentage of reads mapped to Ecoli | | |||
| | %Adapter | Percentage of reads mapped to adapter | | |||
| | %Vector | Percentage of reads mapped to vector | | |||
| | %rRNA | Percentage of reads mapped to rRNA | | |||
| | %Virus | Percentage of reads mapped to virus | | |||
| | %Yeast | Percentage of reads mapped to yeast | | |||
| | %Mitoch | Percentage of reads mapped to mitochondrion | | |||
| | %No hits | Percentage of reads not mapped to genomes mentioned above | | |||
| #### 2. post_alignment.txt | |||
| | Column name | Description | | |||
| | --------------------- | --------------------------------------------- | | |||
| | Sample | Sample name | | |||
| | %Mapping | Percentage of mapped reads | | |||
| | %Mismatch Rate | Mapping error rate | | |||
| | Mendelian Insert Size | Median insert size(bp) | | |||
| | %Q20 | Percentage of bases >Q20 | | |||
| | %Q30 | Percentage of bases >Q30 | | |||
| | Mean Coverage | Mean deduped coverage | | |||
| | Median Coverage | Median deduped coverage | | |||
| | PCT_1X | Fraction of genome with at least 1x coverage | | |||
| | PCT_5X | Fraction of genome with at least 5x coverage | | |||
| | PCT_10X | Fraction of genome with at least 10x coverage | | |||
| | PCT_20X | Fraction of genome with at least 20x coverage | | |||
| | PCT_30X | Fraction of genome with at least 30x coverage | | |||
| | Fold-80 | Fold-80 penalty | | |||
| | On target bases rate | On target bases rate | | |||
| ####3. variants.calling.qc.txt | |||
| | Column name | Description | | |||
| | --------------- | ------------------------------------------------------------ | | |||
| | Sample | Sample name | | |||
| | SNV number | Total SNV number (chr1-22,X) | | |||
| | INDEL number | Total INDEL number (chr1-22,X) | | |||
| | SNV query | SNV number in benchmark region | | |||
| | INDEL query | INDEL number in benchmark region | | |||
| | SNV TP | True positive SNV | | |||
| | INDEL TP | True positive INDEL | | |||
| | SNV FP | False positive SNV | | |||
| | INDEL FP | True positive INDEL | | |||
| | SNV FN | False negative SNV | | |||
| | INDEL FN | False negative INDEL | | |||
| | SNV precision | Precision of SNV calls when compared with benchmark calls in benchmark regions | | |||
| | INDEL precision | Precision of INDEL calls when compared with benchmark calls in benchmark regions | | |||
| | SNV recall | Recall of SNV calls when compared with benchmark calls in benchmark regions | | |||
| | INDEL recall | Recall of INDEL calls when compared with benchmark calls in benchmark regions | | |||
| | SNV F1 | F1 score of SNV calls when compared with benchmark calls in benchmark regions | | |||
| | INDEL F1 | F1 score of INDEL calls when compared with benchmark calls in benchmark regions | | |||
| ####4 {project}.summary.txt | |||
| | Column name | Description | | |||
| | ----------------------------- | ----------------------------------------------------------- | | |||
| | Family | Family name defined by inputed project name | | |||
| | Reproducibility_D5_D6 | Percentage of variants were shared by the twins (D5 and D6) | | |||
| | Mendelian_Concordance_Quartet | Percentage of variants were Mendelian concordance | | |||
BIN
codescripts/.DS_Store
Прегледај датотеку
+ 90
- 0
codescripts/D5_D6.py
Прегледај датотеку
| @@ -0,0 +1,90 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to calculate reproducibility between Quartet_D5 and Quartet_D6s") | |||
| parser.add_argument('-sister', '--sister', type=str, help='sister.txt', required=True) | |||
| parser.add_argument('-project', '--project', type=str, help='project name', required=True) | |||
| args = parser.parse_args() | |||
| sister_file = args.sister | |||
| project_name = args.project | |||
| # output file | |||
| output_name = project_name + '.sister.reproducibility.txt' | |||
| output_file = open(output_name,'w') | |||
| # input files | |||
| sister_dat = pd.read_table(sister_file) | |||
| indel_sister_same = 0 | |||
| indel_sister_diff = 0 | |||
| snv_sister_same = 0 | |||
| snv_sister_diff = 0 | |||
| for row in sister_dat.itertuples(): | |||
| # snv indel | |||
| if ',' in row[4]: | |||
| alt = row[4].split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row[4]) | |||
| alt = alt_max | |||
| ref = row[3] | |||
| if len(ref) == 1 and alt == 1: | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| cate = 'INDEL' | |||
| # sister | |||
| if row[5] == row[6]: | |||
| if row[5] == './.': | |||
| mendelian = 'noInfo' | |||
| sister_count = "no" | |||
| elif row[5] == '0/0': | |||
| mendelian = 'Ref' | |||
| sister_count = "no" | |||
| else: | |||
| mendelian = '1' | |||
| sister_count = "yes_same" | |||
| else: | |||
| mendelian = '0' | |||
| if (row[5] == './.' or row[5] == '0/0') and (row[6] == './.' or row[6] == '0/0'): | |||
| sister_count = "no" | |||
| else: | |||
| sister_count = "yes_diff" | |||
| if cate == 'SNV': | |||
| if sister_count == 'yes_same': | |||
| snv_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| snv_sister_diff += 1 | |||
| else: | |||
| pass | |||
| elif cate == 'INDEL': | |||
| if sister_count == 'yes_same': | |||
| indel_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| indel_sister_diff += 1 | |||
| else: | |||
| pass | |||
| indel_sister = indel_sister_same/(indel_sister_same + indel_sister_diff) | |||
| snv_sister = snv_sister_same/(snv_sister_same + snv_sister_diff) | |||
| outcolumn = 'Project\tReproducibility_D5_D6\n' | |||
| indel_outResult = project_name + '.INDEL' + '\t' + str(indel_sister) + '\n' | |||
| snv_outResult = project_name + '.SNV' + '\t' + str(snv_sister) + '\n' | |||
| output_file.write(outcolumn) | |||
| output_file.write(indel_outResult) | |||
| output_file.write(snv_outResult) | |||
+ 37
- 0
codescripts/Indel_bed.py
Прегледај датотеку
| @@ -0,0 +1,37 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/MIE/vcf/mutation_type',header=None) | |||
| outIndel = open(sys.argv[1],'w') | |||
| for row in mut.itertuples(): | |||
| if ',' in row._4: | |||
| alt_seq = row._4.split(',') | |||
| alt_len = [len(i) for i in alt_seq] | |||
| alt = max(alt_len) | |||
| else: | |||
| alt = len(row._4) | |||
| ref = row._3 | |||
| pos = row._2 | |||
| if len(ref) == 1 and alt == 1: | |||
| pass | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
+ 72
- 0
codescripts/bed_region.py
Прегледај датотеку
| @@ -0,0 +1,72 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = mut = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/vcf/mutation_type',header=None) | |||
| vote = pd.read_table('/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/all_info/benchmark.vote.mendelian.txt',header=None) | |||
| merged_df = pd.merge(vote, mut, how='inner', left_on=[0,1], right_on = [0,1]) | |||
| outFile = open(sys.argv[1],'w') | |||
| outIndel = open(sys.argv[2],'w') | |||
| for row in merged_df.itertuples(): | |||
| #d5 | |||
| if ',' in row._7: | |||
| d5 = row._7.split(',') | |||
| d5_len = [len(i) for i in d5] | |||
| d5_alt = max(d5_len) | |||
| else: | |||
| d5_alt = len(row._7) | |||
| #d6 | |||
| if ',' in row._15: | |||
| d6 = row._15.split(',') | |||
| d6_len = [len(i) for i in d6] | |||
| d6_alt = max(d6_len) | |||
| else: | |||
| d6_alt = len(row._15) | |||
| #f7 | |||
| if ',' in row._23: | |||
| f7 = row._23.split(',') | |||
| f7_len = [len(i) for i in f7] | |||
| f7_alt = max(f7_len) | |||
| else: | |||
| f7_alt = len(row._23) | |||
| #m8 | |||
| if ',' in row._31: | |||
| m8 = row._31.split(',') | |||
| m8_len = [len(i) for i in m8] | |||
| m8_alt = max(m8_len) | |||
| else: | |||
| m8_alt = len(row._31) | |||
| all_length = [d5_alt,d6_alt,f7_alt,m8_alt] | |||
| alt = max(all_length) | |||
| ref = row._35 | |||
| pos = int(row._2) | |||
| if len(ref) == 1 and alt == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline_indel = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\n' | |||
| outIndel.write(outline_indel) | |||
| outline = row._1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\t' + str(row._2) + '\t' + cate + '\n' | |||
| outFile.write(outline) | |||
+ 67
- 0
codescripts/cluster.sh
Прегледај датотеку
| @@ -0,0 +1,67 @@ | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$7"\t.\t.\t.\tGT\t"$6}' | grep -v '0/0' > LCL5.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$15"\t.\t.\t.\tGT\t"$14}' | grep -v '0/0' > LCL6.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$23"\t.\t.\t.\tGT\t"$22}' | grep -v '0/0'> LCL7.body | |||
| cat benchmark.men.vote.diffbed.lengthlessthan50.txt | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$31"\t.\t.\t.\tGT\t"$30}'| grep -v '0/0' > LCL8.body | |||
| cat header5 LCL5.body > LCL5.beforediffbed.vcf | |||
| cat header6 LCL6.body > LCL6.beforediffbed.vcf | |||
| cat header7 LCL7.body > LCL7.beforediffbed.vcf | |||
| cat header8 LCL8.body > LCL8.beforediffbed.vcf | |||
| rtg bgzip *beforediffbed.vcf | |||
| rtg index *beforediffbed.vcf.gz | |||
| rtg vcffilter -i LCL5.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL5.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL6.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL6.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL7.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL7.afterfilterdiffbed.vcf.gz | |||
| rtg vcffilter -i LCL8.beforediffbed.vcf.gz --exclude-bed=/mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed -o LCL8.afterfilterdiffbed.vcf.gz | |||
| /mnt/pgx_src_data_pool_4/home/renluyao/softwares/annovar/table_annovar.pl LCL5.beforediffbed.vcf.gz /mnt/pgx_src_data_pool_4/home/renluyao/softwares/annovar/humandb \ | |||
| -buildver hg38 \ | |||
| -out LCL5 \ | |||
| -remove \ | |||
| -protocol 1000g2015aug_all,1000g2015aug_afr,1000g2015aug_amr,1000g2015aug_eas,1000g2015aug_eur,1000g2015aug_sas,clinvar_20190305,gnomad211_genome \ | |||
| -operation f,f,f,f,f,f,f,f \ | |||
| -nastring . \ | |||
| -vcfinput \ | |||
| --thread 8 | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL5.afterfilterdiffbed.vcf.gz -o LCL5_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL6.afterfilterdiffbed.vcf.gz -o LCL6_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL7.afterfilterdiffbed.vcf.gz -o LCL7_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| rtg vcfeval -b /mnt/pgx_src_data_pool_4/home/renluyao/Quartet/GIAB/NA12878_HG001/HG001_GRCh38_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz -c LCL8.afterfilterdiffbed.vcf.gz -o LCL8_NIST -t /mnt/pgx_src_data_pool_4/home/renluyao/annotation/hg38/GRCh38.d1.vd1.sdf/ | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) == 1)) { print } }' | wc -l | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) < 11) && (length($5) > 1)) { print } }' | wc -l | |||
| zcat LCL5.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL6.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL7.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| zcat LCL8.afterfilterdiffbed.vcf.gz | grep -v '#' | awk '{ if ((length($4) == 1) && (length($5) > 10)) { print } }' | wc -l | |||
| bedtools subtract -a LCL5.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL5.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL6.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL6.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL7.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL7.27.homo_ref.consensus.filtereddiffbed.bed | |||
| bedtools subtract -a LCL8.27.homo_ref.consensus.bed -b /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/MIE/diff.merged.bed > LCL8.27.homo_ref.consensus.filtereddiffbed.bed | |||
| python vcf2bed.py LCL5.body LCL5.variants.bed | |||
| python vcf2bed.py LCL6.body LCL6.variants.bed | |||
| python vcf2bed.py LCL7.body LCL7.variants.bed | |||
| python vcf2bed.py LCL8.body LCL8.variants.bed | |||
| cat /mnt/pgx_src_data_pool_4/home/renluyao/manuscript/benchmark_calls/all_info/LCL5.variants.bed | cut -f1,11,12 | cat - LCL5.27.homo_ref.consensus.filtereddiffbed.bed | sort -k1,1 -k2,2n > LCL5.high.confidence.bed | |||
+ 102
- 0
codescripts/dna_data_summary.py
Прегледај датотеку
| @@ -0,0 +1,102 @@ | |||
| import pandas as pd | |||
| from functools import reduce | |||
| import sys, argparse, os | |||
| import glob | |||
| parser = argparse.ArgumentParser(description="This script is to get information from multiqc and sentieon, output the raw fastq, bam and variants calling (precision and recall) quality metrics") | |||
| parser.add_argument('-pre', '--pre_alignment_path', type=str, help='pre-alignment directory') | |||
| parser.add_argument('-post', '--post_alignment_path', type=str, help='post-alignment directory') | |||
| parser.add_argument('-vcf', '--benchmark_path', type=str, help='benchmark directory') | |||
| parser.add_argument('-men', '--mendelian_path', type=str, help='mendelian directory') | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| pre_alignment_path = args.pre_alignment_path | |||
| post_alignment_path = args.post_alignment_path | |||
| benchmark_path = args.benchmark_path | |||
| mendelian_path = args.mendelian_path | |||
| ###pre-alignment | |||
| pre_alignment_df = pd.concat(map(pd.read_table,glob.glob(os.path.join(pre_alignment_path,'*.txt')))) | |||
| pre_alignment_df.to_csv('pre-alignment-all.txt',sep="\t",index=0) | |||
| ###post-alignment | |||
| post_alignment_df = pd.concat(map(pd.read_table,glob.glob(os.path.join(post_alignment_path,'*.txt')))) | |||
| post_alignment_df.to_csv('post-alignment-all.txt',sep="\t",index=0) | |||
| ###variant-calling qc | |||
| ## detail | |||
| variant_calling_df = pd.concat(map(pd.read_table,glob.glob(os.path.join(benchmark_path,'*.txt')))) | |||
| variant_calling_df.to_csv('variant-calling-all.txt',sep="\t",index=0) | |||
| ## mean + sd | |||
| ####snv | |||
| a = variant_calling_df["SNV precision"].mean().round(2) | |||
| b = variant_calling_df["SNV precision"].std().round(2) | |||
| c = variant_calling_df["SNV recall"].mean().round(2) | |||
| d = variant_calling_df["SNV precision"].std().round(2) | |||
| variant_calling_df["SNV F1 score"] = 2 * variant_calling_df["SNV precision"] * variant_calling_df["SNV recall"] / (variant_calling_df["SNV precision"] + variant_calling_df["SNV recall"]) | |||
| e = variant_calling_df["SNV F1 score"].mean().round(2) | |||
| f = variant_calling_df["SNV F1 score"].std().round(2) | |||
| #### indel | |||
| a2 = variant_calling_df["INDEL precision"].mean().round(2) | |||
| b2 = variant_calling_df["INDEL precision"].std().round(2) | |||
| c2 = variant_calling_df["INDEL recall"].mean().round(2) | |||
| d2 = variant_calling_df["INDEL precision"].std().round(2) | |||
| variant_calling_df["INDEL F1 score"] = 2 * variant_calling_df["INDEL precision"] * variant_calling_df["INDEL recall"] / (variant_calling_df["INDEL precision"] + variant_calling_df["INDEL recall"]) | |||
| e2 = variant_calling_df["INDEL F1 score"].mean().round(2) | |||
| f2 = variant_calling_df["INDEL F1 score"].std().round(2) | |||
| data = {'precision':[str(a),str(a2)], 'precision_sd':[str(b),str(b2)], 'recall':[str(c),str(c2)], 'recall_sd':[str(d),str(d2)], 'F1-score':[str(e),str(e2)], 'F1-score_sd':[str(f),str(f2)] } | |||
| df = pd.DataFrame(data, index =['SNV', 'INDEL']) | |||
| df.to_csv('benchmark_summary.txt',sep="\t") | |||
| ### Mendelian | |||
| mendelian_df = pd.concat(map(pd.read_table,glob.glob(os.path.join(mendelian_path,'*.txt')))) | |||
| mendelian_df.to_csv('mendelian-all.txt',sep="\t",index=0) | |||
| ### snv | |||
| snv = mendelian_df[mendelian_df['Family'].str.contains("SNV")] | |||
| indel = mendelian_df[mendelian_df['Family'].str.contains("INDEL")] | |||
| g = snv["Mendelian_Concordance_Rate"].mean().round(2) | |||
| h = snv["Mendelian_Concordance_Rate"].std().round(2) | |||
| g2 = indel["Mendelian_Concordance_Rate"].mean().round(2) | |||
| h2 = indel["Mendelian_Concordance_Rate"].std().round(2) | |||
| data = {'MCR':[str(g),str(g2)], 'MCR_sd':[str(h),str(h2)]} | |||
| df = pd.DataFrame(data, index =['SNV', 'INDEL']) | |||
| df.to_csv('mendelian_summary.txt',sep="\t") | |||
+ 137
- 0
codescripts/extract_tables.py
Прегледај датотеку
| @@ -0,0 +1,137 @@ | |||
| import json | |||
| import pandas as pd | |||
| from functools import reduce | |||
| import sys, argparse, os | |||
| parser = argparse.ArgumentParser(description="This script is to get information from multiqc and sentieon, output the raw fastq, bam and variants calling (precision and recall) quality metrics") | |||
| parser.add_argument('-quality', '--quality_yield', type=str, help='*.quality_yield.txt') | |||
| parser.add_argument('-depth', '--wgs_metrics', type=str, help='*deduped_WgsMetricsAlgo.txt') | |||
| parser.add_argument('-aln', '--aln_metrics', type=str, help='*_deduped_aln_metrics.txt') | |||
| parser.add_argument('-is', '--is_metrics', type=str, help='*_deduped_is_metrics.txt') | |||
| parser.add_argument('-hs', '--hs_metrics', type=str, help='*_deduped_hs_metrics.txt') | |||
| parser.add_argument('-fastqc', '--fastqc', type=str, help='multiqc_fastqc.txt') | |||
| parser.add_argument('-fastqscreen', '--fastqscreen', type=str, help='multiqc_fastq_screen.txt') | |||
| parser.add_argument('-hap', '--happy', type=str, help='multiqc_happy_data.json', required=True) | |||
| parser.add_argument('-project', '--project_name', type=str, help='project_name') | |||
| args = parser.parse_args() | |||
| if args.quality_yield: | |||
| # Rename input: | |||
| quality_yield_file = args.quality_yield | |||
| wgs_metrics_file = args.wgs_metrics | |||
| aln_metrics_file = args.aln_metrics | |||
| is_metrics_file = args.is_metrics | |||
| fastqc_file = args.fastqc | |||
| fastqscreen_file = args.fastqscreen | |||
| hap_file = args.happy | |||
| project_name = args.project_name | |||
| ############################################# | |||
| # fastqc | |||
| fastqc = pd.read_table(fastqc_file) | |||
| # fastqscreen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| dat['Sample'] = [i.replace('_screen','') for i in dat['Sample']] | |||
| fastqscreen.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| # pre-alignment | |||
| pre_alignment_dat = pd.merge(fastqc,fastqscreen,how="outer",left_on=['Sample'],right_on=['Sample']) | |||
| pre_alignment_dat['FastQC_mqc-generalstats-fastqc-total_sequences'] = pre_alignment_dat['FastQC_mqc-generalstats-fastqc-total_sequences']/1000000 | |||
| del pre_alignment_dat['FastQC_mqc-generalstats-fastqc-percent_fails'] | |||
| del pre_alignment_dat['FastQC_mqc-generalstats-fastqc-avg_sequence_length'] | |||
| del pre_alignment_dat['ERCC percentage'] | |||
| del pre_alignment_dat['Phix percentage'] | |||
| del pre_alignment_dat['Mouse percentage'] | |||
| pre_alignment_dat = pre_alignment_dat.round(2) | |||
| pre_alignment_dat.columns = ['Sample','%Dup','%GC','Total Sequences (million)','%Human','%EColi','%Adapter','%Vector','%rRNA','%Virus','%Yeast','%Mitoch','%No hits'] | |||
| pre_alignment_dat.to_csv('pre_alignment.txt',sep="\t",index=0) | |||
| ############################ | |||
| dat = pd.read_table(aln_metrics_file,index_col=False) | |||
| dat['PCT_ALIGNED_READS'] = dat["PF_READS_ALIGNED"]/dat["TOTAL_READS"] | |||
| aln_metrics = dat[["Sample", "PCT_ALIGNED_READS","PF_MISMATCH_RATE"]] | |||
| aln_metrics = aln_metrics * 100 | |||
| aln_metrics['Sample'] = [x[-1] for x in aln_metrics['Sample'].str.split('/')] | |||
| dat = pd.read_table(is_metrics_file,index_col=False) | |||
| is_metrics = dat[['Sample', 'MEDIAN_INSERT_SIZE']] | |||
| is_metrics['Sample'] = [x[-1] for x in is_metrics['Sample'].str.split('/')] | |||
| dat = pd.read_table(quality_yield_file,index_col=False) | |||
| dat['%Q20'] = dat['Q20_BASES']/dat['TOTAL_BASES'] | |||
| dat['%Q30'] = dat['Q30_BASES']/dat['TOTAL_BASES'] | |||
| quality_yield = dat[['Sample','%Q20','%Q30']] | |||
| quality_yield = quality_yield * 100 | |||
| quality_yield['Sample'] = [x[-1] for x in quality_yield['Sample'].str.split('/')] | |||
| dat = pd.read_table(wgs_metrics_file,index_col=False) | |||
| wgs_metrics = dat[['Sample','MEDIAN_COVERAGE','PCT_1X', 'PCT_5X', 'PCT_10X','PCT_20X','PCT_30X']] | |||
| wgs_metrics['PCT_1X'] = wgs_metrics['PCT_1X'] * 100 | |||
| wgs_metrics['PCT_5X'] = wgs_metrics['PCT_5X'] * 100 | |||
| wgs_metrics['PCT_10X'] = wgs_metrics['PCT_10X'] * 100 | |||
| wgs_metrics['PCT_20X'] = wgs_metrics['PCT_20X'] * 100 | |||
| wgs_metrics['PCT_30X'] = wgs_metrics['PCT_30X'] * 100 | |||
| wgs_metrics['Sample'] = [x[-1] for x in wgs_metrics['Sample'].str.split('/')] | |||
| hs_metrics = dat[['Sample','FOLD_80_BASE_PENALTY','PCT_USABLE_BASES_ON_TARGET']] | |||
| data_frames = [aln_metrics, is_metrics, quality_yield, wgs_metrics, hs_metrics] | |||
| post_alignment_dat = reduce(lambda left,right: pd.merge(left,right,on=['Sample'],how='outer'), data_frames) | |||
| post_alignment_dat.columns = ['Sample', '%Mapping', '%Mismatch Rate', 'Mendelian Insert Size','%Q20', '%Q30', 'Median Coverage', 'PCT_1X', 'PCT_5X', 'PCT_10X','PCT_20X','PCT_30X','Fold-80','On target bases rate'] | |||
| post_alignment_dat = post_alignment_dat.round(2) | |||
| post_alignment_dat.to_csv('post_alignment.txt',sep="\t",index=0) | |||
| ######################################### | |||
| # variants calling | |||
| with open(hap_file) as hap_json: | |||
| happy = json.load(hap_json) | |||
| dat =pd.DataFrame.from_records(happy) | |||
| dat = dat.loc[:, dat.columns.str.endswith('ALL')] | |||
| dat_transposed = dat.T | |||
| dat_transposed = dat_transposed.loc[:,['sample_id','QUERY.TOTAL','METRIC.Precision','METRIC.Recall']] | |||
| indel = dat_transposed[['INDEL' in s for s in dat_transposed.index]] | |||
| snv = dat_transposed[['SNP' in s for s in dat_transposed.index]] | |||
| indel.reset_index(drop=True, inplace=True) | |||
| snv.reset_index(drop=True, inplace=True) | |||
| benchmark = pd.concat([snv, indel], axis=1) | |||
| benchmark = benchmark[["sample_id", 'QUERY.TOTAL', 'METRIC.Precision', 'METRIC.Recall']] | |||
| benchmark.columns = ['Sample','sample_id','SNV number','INDEL number','SNV precision','INDEL precision','SNV recall','INDEL recall'] | |||
| benchmark = benchmark[['Sample','SNV number','INDEL number','SNV precision','INDEL precision','SNV recall','INDEL recall']] | |||
| benchmark['SNV precision'] = benchmark['SNV precision'].astype(float) | |||
| benchmark['INDEL precision'] = benchmark['INDEL precision'].astype(float) | |||
| benchmark['SNV recall'] = benchmark['SNV recall'].astype(float) | |||
| benchmark['INDEL recall'] = benchmark['INDEL recall'].astype(float) | |||
| benchmark['SNV precision'] = benchmark['SNV precision'] * 100 | |||
| benchmark['INDEL precision'] = benchmark['INDEL precision'] * 100 | |||
| benchmark['SNV recall'] = benchmark['SNV recall'] * 100 | |||
| benchmark['INDEL recall'] = benchmark['INDEL recall']* 100 | |||
| benchmark = benchmark.round(2) | |||
| benchmark.to_csv('variants.calling.qc.txt',sep="\t",index=0) | |||
| else: | |||
| hap_file = args.happy | |||
| with open(hap_file) as hap_json: | |||
| happy = json.load(hap_json) | |||
| dat =pd.DataFrame.from_records(happy) | |||
| dat = dat.loc[:, dat.columns.str.endswith('ALL')] | |||
| dat_transposed = dat.T | |||
| dat_transposed = dat_transposed.loc[:,['sample_id','QUERY.TOTAL','METRIC.Precision','METRIC.Recall']] | |||
| indel = dat_transposed[['INDEL' in s for s in dat_transposed.index]] | |||
| snv = dat_transposed[['SNP' in s for s in dat_transposed.index]] | |||
| indel.reset_index(drop=True, inplace=True) | |||
| snv.reset_index(drop=True, inplace=True) | |||
| benchmark = pd.concat([snv, indel], axis=1) | |||
| benchmark = benchmark[["sample_id", 'QUERY.TOTAL', 'METRIC.Precision', 'METRIC.Recall']] | |||
| benchmark.columns = ['Sample','sample_id','SNV number','INDEL number','SNV precision','INDEL precision','SNV recall','INDEL recall'] | |||
| benchmark = benchmark[['Sample','SNV number','INDEL number','SNV precision','INDEL precision','SNV recall','INDEL recall']] | |||
| benchmark['SNV precision'] = benchmark['SNV precision'].astype(float) | |||
| benchmark['INDEL precision'] = benchmark['INDEL precision'].astype(float) | |||
| benchmark['SNV recall'] = benchmark['SNV recall'].astype(float) | |||
| benchmark['INDEL recall'] = benchmark['INDEL recall'].astype(float) | |||
| benchmark['SNV precision'] = benchmark['SNV precision'] * 100 | |||
| benchmark['INDEL precision'] = benchmark['INDEL precision'] * 100 | |||
| benchmark['SNV recall'] = benchmark['SNV recall'] * 100 | |||
| benchmark['INDEL recall'] = benchmark['INDEL recall']* 100 | |||
| benchmark = benchmark.round(2) | |||
| benchmark.to_csv('variants.calling.qc.txt',sep="\t",index=0) | |||
+ 92
- 0
codescripts/extract_vcf_information.py
Прегледај датотеку
| @@ -0,0 +1,92 @@ | |||
| import sys,getopt | |||
| import os | |||
| import re | |||
| import fileinput | |||
| import pandas as pd | |||
| def usage(): | |||
| print( | |||
| """ | |||
| Usage: python extract_vcf_information.py -i input_merged_vcf_file -o parsed_file | |||
| This script will extract SNVs and Indels information from the vcf files and output a tab-delimited files. | |||
| Input: | |||
| -i the selected vcf file | |||
| Output: | |||
| -o tab-delimited parsed file | |||
| """) | |||
| # select supported small variants | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| infoParsed = parse_INFO(strings[7]) | |||
| formatKeys = strings[8].split(':') | |||
| formatValues = strings[9].split(':') | |||
| for i in range(0,len(formatKeys) -1) : | |||
| if formatKeys[i] == 'AD': | |||
| ra = formatValues[i].split(',') | |||
| infoParsed['RefDP'] = ra[0] | |||
| infoParsed['AltDP'] = ra[1] | |||
| if (int(ra[1]) + int(ra[0])) != 0: | |||
| infoParsed['af'] = float(int(ra[1])/(int(ra[1]) + int(ra[0]))) | |||
| else: | |||
| pass | |||
| else: | |||
| infoParsed[formatKeys[i]] = formatValues[i] | |||
| infoParsed['chromo'] = strings[0] | |||
| infoParsed['pos'] = strings[1] | |||
| infoParsed['id'] = strings[2] | |||
| infoParsed['ref'] = strings[3] | |||
| infoParsed['alt'] = strings[4] | |||
| infoParsed['qual'] = strings[5] | |||
| return infoParsed | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| elif kv[0] == 'AF': | |||
| pass | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| opts,args = getopt.getopt(sys.argv[1:],"hi:o:") | |||
| for op,value in opts: | |||
| if op == "-i": | |||
| inputFile=value | |||
| elif op == "-o": | |||
| outputFile=value | |||
| elif op == "-h": | |||
| usage() | |||
| sys.exit() | |||
| if len(sys.argv[1:]) < 3: | |||
| usage() | |||
| sys.exit() | |||
| allDict = [] | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| oneDict = process(line) | |||
| allDict.append(oneDict) | |||
| allTable = pd.DataFrame(allDict) | |||
| allTable.to_csv(outputFile,sep='\t',index=False) | |||
+ 47
- 0
codescripts/filter_indel_over_50_cluster.py
Прегледај датотеку
| @@ -0,0 +1,47 @@ | |||
| import sys,getopt | |||
| from itertools import islice | |||
| over_50_outfile = open("indel_lenth_over_50.txt",'w') | |||
| less_50_outfile = open("benchmark.men.vote.diffbed.lengthlessthan50.txt","w") | |||
| def process(line): | |||
| strings = line.strip().split('\t') | |||
| #d5 | |||
| if ',' in strings[6]: | |||
| d5 = strings[6].split(',') | |||
| d5_len = [len(i) for i in d5] | |||
| d5_alt = max(d5_len) | |||
| else: | |||
| d5_alt = len(strings[6]) | |||
| #d6 | |||
| if ',' in strings[14]: | |||
| d6 = strings[14].split(',') | |||
| d6_len = [len(i) for i in d6] | |||
| d6_alt = max(d6_len) | |||
| else: | |||
| d6_alt = len(strings[14]) | |||
| #f7 | |||
| if ',' in strings[22]: | |||
| f7 = strings[22].split(',') | |||
| f7_len = [len(i) for i in f7] | |||
| f7_alt = max(f7_len) | |||
| else: | |||
| f7_alt = len(strings[22]) | |||
| #m8 | |||
| if ',' in strings[30]: | |||
| m8 = strings[30].split(',') | |||
| m8_len = [len(i) for i in m8] | |||
| m8_alt = max(m8_len) | |||
| else: | |||
| m8_alt = len(strings[30]) | |||
| #ref | |||
| ref_len = len(strings[34]) | |||
| if (d5_alt > 50) or (d6_alt > 50) or (f7_alt > 50) or (m8_alt > 50) or (ref_len > 50): | |||
| over_50_outfile.write(line) | |||
| else: | |||
| less_50_outfile.write(line) | |||
| input_file = open(sys.argv[1]) | |||
| for line in islice(input_file, 1, None): | |||
| process(line) | |||
+ 43
- 0
codescripts/filter_indel_over_50_mendelian.py
Прегледај датотеку
| @@ -0,0 +1,43 @@ | |||
| from itertools import islice | |||
| import fileinput | |||
| import sys, argparse, os | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to exclude indel over 50bp") | |||
| parser.add_argument('-i', '--mergedGVCF', type=str, help='merged gVCF txt with only chr, pos, ref, alt and genotypes', required=True) | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='prefix of output file', required=True) | |||
| args = parser.parse_args() | |||
| input_dat = args.mergedGVCF | |||
| prefix = args.prefix | |||
| # output file | |||
| output_name = prefix + '.indel.lessthan50bp.txt' | |||
| outfile = open(output_name,'w') | |||
| def process(line): | |||
| strings = line.strip().split('\t') | |||
| #d5 | |||
| if ',' in strings[3]: | |||
| alt = strings[3].split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(strings[3]) | |||
| #ref | |||
| ref_len = len(strings[2]) | |||
| if (alt_max > 50) or (ref_len > 50): | |||
| pass | |||
| else: | |||
| outfile.write(line) | |||
| for line in fileinput.input(input_dat): | |||
| process(line) | |||
+ 416
- 0
codescripts/high_confidence_call_vote.py
Прегледај датотеку
| @@ -0,0 +1,416 @@ | |||
| from __future__ import division | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to count voting number") | |||
| parser.add_argument('-vcf', '--multi_sample_vcf', type=str, help='The VCF file you want to count the voting number', required=True) | |||
| parser.add_argument('-dup', '--dup_list', type=str, help='Duplication list', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='Prefix of output file name', required=True) | |||
| args = parser.parse_args() | |||
| multi_sample_vcf = args.multi_sample_vcf | |||
| dup_list = args.dup_list | |||
| prefix = args.prefix | |||
| sample_name = args.sample_name | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Sum depth of all samples"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Sum alternative depth of all samples"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency, sum alternative depth / sum depth"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Average genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Average mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUALI,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| vcf_header_all_sample = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=location,Number=1,Type=String,Description="Repeat region"> | |||
| ##INFO=<ID=DUP,Number=1,Type=Flag,Description="Duplicated variant records"> | |||
| ##INFO=<ID=DETECTED,Number=1,Type=Integer,Description="Number of detected votes"> | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number of consnesus votes"> | |||
| ##INFO=<ID=FAM,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##INFO=<ID=ALL_ALT,Number=1,Type=Float,Description="Sum of alternative reads of all samples"> | |||
| ##INFO=<ID=ALL_DP,Number=1,Type=Float,Description="Sum of depth of all samples"> | |||
| ##INFO=<ID=ALL_AF,Number=1,Type=Float,Description="Allele frequency of net alternatice reads and net depth"> | |||
| ##INFO=<ID=GQ_MEAN,Number=1,Type=Float,Description="Mean of genotype quality of all samples"> | |||
| ##INFO=<ID=QD_MEAN,Number=1,Type=Float,Description="Average Variant Confidence/Quality by Depth"> | |||
| ##INFO=<ID=MQ_MEAN,Number=1,Type=Float,Description="Mean of mapping quality of all samples"> | |||
| ##INFO=<ID=FS_MEAN,Number=1,Type=Float,Description="Average Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##INFO=<ID=QUAL_MEAN,Number=1,Type=Float,Description="Average variant quality"> | |||
| ##INFO=<ID=PCR,Number=1,Type=String,Description="Consensus of PCR votes"> | |||
| ##INFO=<ID=PCR_FREE,Number=1,Type=String,Description="Consensus of PCR-free votes"> | |||
| ##INFO=<ID=CONSENSUS,Number=1,Type=String,Description="Consensus calls"> | |||
| ##INFO=<ID=CONSENSUS_SEQ,Number=1,Type=String,Description="Consensus sequence"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=String,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=String,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=String,Description="Genotype quality"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=String,Description="Mapping quality"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=String,Description="1 is twins shared, 0 is twins discordant "> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL5 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=String,Description="1 is LCL7, LCL8 and LCL6 mendelian consistent, 0 is mendelian vioaltion"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # read in duplication list | |||
| dup = pd.read_table(dup_list,header=None) | |||
| var_dup = dup[0].tolist() | |||
| # output file | |||
| benchmark_file_name = prefix + '_voted.vcf' | |||
| benchmark_outfile = open(benchmark_file_name,'w') | |||
| all_sample_file_name = prefix + '_all_sample_information.vcf' | |||
| all_sample_outfile = open(all_sample_file_name,'w') | |||
| # write VCF | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '_benchmark_calls\n' | |||
| benchmark_outfile.write(vcf_header) | |||
| benchmark_outfile.write(outputcolumn) | |||
| outputcolumn_all_sample = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+ \ | |||
| 'Quartet_DNA_BGI_SEQ2000_BGI_1_20180518\tQuartet_DNA_BGI_SEQ2000_BGI_2_20180530\tQuartet_DNA_BGI_SEQ2000_BGI_3_20180530\t' + \ | |||
| 'Quartet_DNA_BGI_T7_WGE_1_20191105\tQuartet_DNA_BGI_T7_WGE_2_20191105\tQuartet_DNA_BGI_T7_WGE_3_20191105\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_1_20181108\tQuartet_DNA_ILM_Nova_ARD_2_20181108\tQuartet_DNA_ILM_Nova_ARD_3_20181108\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_ARD_4_20190111\tQuartet_DNA_ILM_Nova_ARD_5_20190111\tQuartet_DNA_ILM_Nova_ARD_6_20190111\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_BRG_1_20180930\tQuartet_DNA_ILM_Nova_BRG_2_20180930\tQuartet_DNA_ILM_Nova_BRG_3_20180930\t' + \ | |||
| 'Quartet_DNA_ILM_Nova_WUX_1_20190917\tQuartet_DNA_ILM_Nova_WUX_2_20190917\tQuartet_DNA_ILM_Nova_WUX_3_20190917\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_ARD_1_20170403\tQuartet_DNA_ILM_XTen_ARD_2_20170403\tQuartet_DNA_ILM_XTen_ARD_3_20170403\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_NVG_1_20170329\tQuartet_DNA_ILM_XTen_NVG_2_20170329\tQuartet_DNA_ILM_XTen_NVG_3_20170329\t' + \ | |||
| 'Quartet_DNA_ILM_XTen_WUX_1_20170216\tQuartet_DNA_ILM_XTen_WUX_2_20170216\tQuartet_DNA_ILM_XTen_WUX_3_20170216\n' | |||
| all_sample_outfile.write(vcf_header_all_sample) | |||
| all_sample_outfile.write(outputcolumn_all_sample) | |||
| #function | |||
| def replace_nan(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| updated_list.append('.:.:.:.:.:.:.:.:.:.:.:.') | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def remove_dot(strings_list): | |||
| updated_list = [] | |||
| for i in strings_list: | |||
| if i == '.': | |||
| pass | |||
| else: | |||
| updated_list.append(i) | |||
| return updated_list | |||
| def detected_number(strings): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| percentage = 27 - gt.count('.') | |||
| return(str(percentage)) | |||
| def vote_number(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| vote_num = gt.count(consensus_call) | |||
| return(str(vote_num)) | |||
| def family_vote(strings,consensus_call): | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| indices = [i for i, x in enumerate(gt) if x == consensus_call] | |||
| matched_mendelian = itemgetter(*indices)(mendelian) | |||
| mendelian_num = matched_mendelian.count('1:1:1') | |||
| return(str(mendelian_num)) | |||
| def gt_uniform(strings): | |||
| uniformed_gt = '' | |||
| allele1 = strings.split('/')[0] | |||
| allele2 = strings.split('/')[1] | |||
| if int(allele1) > int(allele2): | |||
| uniformed_gt = allele2 + '/' + allele1 | |||
| else: | |||
| uniformed_gt = allele1 + '/' + allele2 | |||
| return uniformed_gt | |||
| def decide_by_rep(strings): | |||
| consensus_rep = '' | |||
| mendelian = [':'.join(x.split(':')[1:4]) for x in strings] | |||
| gt = [x.split(':')[0] for x in strings] | |||
| gt = [x.replace('.','0/0') for x in gt] | |||
| # modified gt turn 2/1 to 1/2 | |||
| gt = list(map(gt_uniform,[i for i in gt])) | |||
| # mendelian consistent? | |||
| mendelian_dict = Counter(mendelian) | |||
| highest_mendelian = mendelian_dict.most_common(1) | |||
| candidate_mendelian = highest_mendelian[0][0] | |||
| freq_mendelian = highest_mendelian[0][1] | |||
| if (candidate_mendelian == '1:1:1') and (freq_mendelian >= 2): | |||
| gt_num_dict = Counter(gt) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if (candidate_gt != '0/0') and (freq_gt >= 2): | |||
| consensus_rep = candidate_gt | |||
| elif (candidate_gt == '0/0') and (freq_gt >= 2): | |||
| consensus_rep = '0/0' | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| elif (candidate_mendelian == '') and (freq_mendelian >= 2): | |||
| consensus_rep = 'noInfo' | |||
| else: | |||
| consensus_rep = 'inconMen' | |||
| return consensus_rep | |||
| def main(): | |||
| for line in fileinput.input(multi_sample_vcf): | |||
| headline = re.match('^\#',line) | |||
| if headline is not None: | |||
| pass | |||
| else: | |||
| line = line.strip() | |||
| strings = line.split('\t') | |||
| variant_id = '_'.join([strings[0],strings[1]]) | |||
| # check if the variants location is duplicated | |||
| if variant_id in var_dup: | |||
| strings[7] = strings[7] + ';DUP' | |||
| outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(outLine) | |||
| else: | |||
| # pre-define | |||
| pcr_consensus = '.' | |||
| pcr_free_consensus = '.' | |||
| consensus_call = '.' | |||
| consensus_alt_seq = '.' | |||
| # pcr | |||
| strings[9:] = replace_nan(strings[9:]) | |||
| pcr = itemgetter(*[9,10,11,27,28,29,30,31,32,33,34,35])(strings) | |||
| SEQ2000 = decide_by_rep(pcr[0:3]) | |||
| XTen_ARD = decide_by_rep(pcr[3:6]) | |||
| XTen_NVG = decide_by_rep(pcr[6:9]) | |||
| XTen_WUX = decide_by_rep(pcr[9:12]) | |||
| sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 2: | |||
| pcr_consensus = candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| pcr_free = itemgetter(*[12,13,14,15,16,17,18,19,20,21,22,23,24,25,26])(strings) | |||
| T7_WGE = decide_by_rep(pcr_free[0:3]) | |||
| Nova_ARD_1 = decide_by_rep(pcr_free[3:6]) | |||
| Nova_ARD_2 = decide_by_rep(pcr_free[6:9]) | |||
| Nova_BRG = decide_by_rep(pcr_free[9:12]) | |||
| Nova_WUX = decide_by_rep(pcr_free[12:15]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG,Nova_WUX] | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 3: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| # pcr and pcr-free | |||
| tag = ['inconGT','noInfo','inconMen','inconSequenceSite'] | |||
| if (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag) and (pcr_consensus != '0/0'): | |||
| consensus_call = pcr_consensus | |||
| VOTED = vote_number(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';VOTED=' + VOTED | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| FAM = family_vote(strings[9:],consensus_call) | |||
| strings[7] = strings[7] + ';FAM=' + FAM | |||
| # Delete multiple alternative genotype to necessary expression | |||
| alt = strings[4] | |||
| alt_gt = alt.split(',') | |||
| if len(alt_gt) > 1: | |||
| allele1 = consensus_call.split('/')[0] | |||
| allele2 = consensus_call.split('/')[1] | |||
| if allele1 == '0': | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| consensus_alt_seq = allele2_seq | |||
| consensus_call = '0/1' | |||
| else: | |||
| allele1_seq = alt_gt[int(allele1) - 1] | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| if int(allele1) > int(allele2): | |||
| consensus_alt_seq = allele2_seq + ',' + allele1_seq | |||
| consensus_call = '1/2' | |||
| elif int(allele1) < int(allele2): | |||
| consensus_alt_seq = allele1_seq + ',' + allele2_seq | |||
| consensus_call = '1/2' | |||
| else: | |||
| consensus_alt_seq = allele1_seq | |||
| consensus_call = '1/1' | |||
| else: | |||
| consensus_alt_seq = alt | |||
| # GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL | |||
| # GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT | |||
| # DP | |||
| DP = [x.split(':')[4] for x in strings[9:]] | |||
| DP = remove_dot(DP) | |||
| DP = [int(x) for x in DP] | |||
| ALL_DP = sum(DP) | |||
| # AF | |||
| ALT = [x.split(':')[5] for x in strings[9:]] | |||
| ALT = remove_dot(ALT) | |||
| ALT = [int(x) for x in ALT] | |||
| ALL_ALT = sum(ALT) | |||
| ALL_AF = round(ALL_ALT/ALL_DP,2) | |||
| # GQ | |||
| GQ = [x.split(':')[7] for x in strings[9:]] | |||
| GQ = remove_dot(GQ) | |||
| GQ = [int(x) for x in GQ] | |||
| GQ_MEAN = round(mean(GQ),2) | |||
| # QD | |||
| QD = [x.split(':')[8] for x in strings[9:]] | |||
| QD = remove_dot(QD) | |||
| QD = [float(x) for x in QD] | |||
| QD_MEAN = round(mean(QD),2) | |||
| # MQ | |||
| MQ = [x.split(':')[9] for x in strings[9:]] | |||
| MQ = remove_dot(MQ) | |||
| MQ = [float(x) for x in MQ] | |||
| MQ_MEAN = round(mean(MQ),2) | |||
| # FS | |||
| FS = [x.split(':')[10] for x in strings[9:]] | |||
| FS = remove_dot(FS) | |||
| FS = [float(x) for x in FS] | |||
| FS_MEAN = round(mean(FS),2) | |||
| # QUAL | |||
| QUAL = [x.split(':')[11] for x in strings[9:]] | |||
| QUAL = remove_dot(QUAL) | |||
| QUAL = [float(x) for x in QUAL] | |||
| QUAL_MEAN = round(mean(QUAL),2) | |||
| # benchmark output | |||
| output_format = consensus_call + ':' + str(ALL_DP) + ':' + str(ALL_ALT) + ':' + str(ALL_AF) + ':' + str(GQ_MEAN) + ':' + str(QD_MEAN) + ':' + str(MQ_MEAN) + ':' + str(FS_MEAN) + ':' + str(QUAL_MEAN) | |||
| outLine = strings[0] + '\t' + strings[1] + '\t' + strings[2] + '\t' + strings[3] + '\t' + consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' + strings[7] + '\t' + 'GT:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + output_format + '\n' | |||
| benchmark_outfile.write(outLine) | |||
| # all sample output | |||
| strings[7] = strings[7] + ';ALL_ALT=' + str(ALL_ALT) + ';ALL_DP=' + str(ALL_DP) + ';ALL_AF=' + str(ALL_AF) \ | |||
| + ';GQ_MEAN=' + str(GQ_MEAN) + ';QD_MEAN=' + str(QD_MEAN) + ';MQ_MEAN=' + str(MQ_MEAN) + ';FS_MEAN=' + str(FS_MEAN) \ | |||
| + ';QUAL_MEAN=' + str(QUAL_MEAN) + ';PCR=' + consensus_call + ';PCR_FREE=' + consensus_call + ';CONSENSUS=' + consensus_call \ | |||
| + ';CONSENSUS_SEQ=' + consensus_alt_seq | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus == '0/0') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != '0/0')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif ((pcr_consensus != '0/0') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == '0/0')): | |||
| consensus_call = 'pcr-speicifc' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call + ';PCR=' + pcr_consensus + ';PCR_FREE=' + pcr_free_consensus | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = 'confirm for parents' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| else: | |||
| consensus_call = 'filtered' | |||
| DETECTED = detected_number(strings[9:]) | |||
| strings[7] = strings[7] + ';DETECTED=' + DETECTED | |||
| strings[7] = strings[7] + ';CONSENSUS=' + consensus_call | |||
| all_sample_outLine = '\t'.join(strings) + '\n' | |||
| all_sample_outfile.write(all_sample_outLine) | |||
| if __name__ == '__main__': | |||
| main() | |||
+ 42
- 0
codescripts/high_voted_mendelian_bed.py
Прегледај датотеку
| @@ -0,0 +1,42 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| mut = pd.read_table(sys.argv[1]) | |||
| outFile = open(sys.argv[2],'w') | |||
| for row in mut.itertuples(): | |||
| #d5 | |||
| if ',' in row.V4: | |||
| alt = row.V4.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.V4) | |||
| #d6 | |||
| alt = alt_max | |||
| ref = row.V3 | |||
| pos = int(row.V2) | |||
| if len(ref) == 1 and alt == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (alt - 1) | |||
| cate = 'INDEL' | |||
| outline = row.V1 + '\t' + str(StartPos) + '\t' + str(EndPos) + '\t' + str(row.V2) + '\t' + cate + '\n' | |||
| outFile.write(outline) | |||
+ 50
- 0
codescripts/how_many_samples.py
Прегледај датотеку
| @@ -0,0 +1,50 @@ | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| from operator import itemgetter | |||
| parser = argparse.ArgumentParser(description="This script is to get how many samples") | |||
| parser.add_argument('-sample', '--sample', type=str, help='quartet_sample', required=True) | |||
| parser.add_argument('-rep', '--rep', type=str, help='quartet_rep', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| sample = args.sample | |||
| rep = args.rep | |||
| quartet_sample = pd.read_table(sample,header=None) | |||
| quartet_sample = list(quartet_sample[0]) | |||
| quartet_rep = pd.read_table(rep.header=None) | |||
| quartet_rep = quartet_rep[0] | |||
| #tags | |||
| sister_tag = 'false' | |||
| quartet_tag = 'false' | |||
| quartet_rep_unique = list(set(quartet_rep)) | |||
| single_rep = [i for i in range(len(quartet_rep)) if quartet_rep[i] == quartet_rep_unique[0]] | |||
| single_batch_sample = itemgetter(*single_rep)(quartet_sample) | |||
| num = len(single_batch_sample) | |||
| if num == 1: | |||
| sister_tag = 'false' | |||
| quartet_tag = 'false' | |||
| elif num == 2: | |||
| if set(single_batch_sample) == set(['LCL5','LCL6']): | |||
| sister_tag = 'true' | |||
| quartet_tag = 'false' | |||
| elif num == 3: | |||
| if ('LCL5' in single_batch_sample) and ('LCL6' in single_batch_sample): | |||
| sister_tag = 'true' | |||
| quartet_tag = 'false' | |||
| elif num == 4: | |||
| if set(single_batch_sample) == set(['LCL5','LCL6','LCL7','LCL8']): | |||
| sister_tag = 'false' | |||
| quartet_tag = 'true' | |||
| sister_outfile = open('sister_tag','w') | |||
| quartet_outfile = open('quartet_tag','w') | |||
| sister_outfile.write(sister_tag) | |||
| quartet_outfile.write(quartet_tag) | |||
+ 42
- 0
codescripts/lcl5_all_called_variants.py
Прегледај датотеку
| @@ -0,0 +1,42 @@ | |||
| from __future__ import division | |||
| import sys, argparse, os | |||
| import pandas as pd | |||
| from collections import Counter | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| vcf = args.vcf | |||
| lcl5_outfile = open('LCL5_all_variants.txt','w') | |||
| filtered_outfile = open('LCL5_filtered_variants.txt','w') | |||
| vcf_dat = pd.read_table(vcf) | |||
| for row in vcf_dat.itertuples(): | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl5_vcf_gt = [x.split(':')[0] for x in lcl5_list] | |||
| lcl5_gt=[item.replace('./.', '0/0') for item in lcl5_vcf_gt] | |||
| gt_dict = Counter(lcl5_gt) | |||
| highest_gt = gt_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| output = row._1 + '\t' + str(row.POS) + '\t' + '\t'.join(lcl5_gt) + '\n' | |||
| if (candidate_gt == '0/0') and (freq_gt == 27): | |||
| filtered_outfile.write(output) | |||
| else: | |||
| lcl5_outfile.write(output) | |||
+ 6
- 0
codescripts/linux_command.sh
Прегледај датотеку
| @@ -0,0 +1,6 @@ | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$7"\t.\t.\t.\tGT\t"$6}' | grep -v '2_y' > LCL5.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$15"\t.\t.\t.\tGT\t"$14}' | grep -v '2_y' > LCL6.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$23"\t.\t.\t.\tGT\t"$22}' | grep -v '2_y' > LCL7.body | |||
| cat benchmark.men.vote.diffbed.filtered | awk '{print $1"\t"$2"\t"".""\t"$35"\t"$31"\t.\t.\t.\tGT\t"$30}' | grep -v '2_y' > LCL8.body | |||
| for i in *txt; do cat $i | awk '{ if ((length($3) == 1) && (length($4) == 1)) { print } }' | grep -v '#' | cut -f3,4 | sort |uniq -c | sed 's/\s\+/\t/g' | cut -f2 > $i.mut; done | |||
+ 129
- 0
codescripts/merge_mendelian_vcfinfo.py
Прегледај датотеку
| @@ -0,0 +1,129 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to get final high confidence calls and information of all replicates") | |||
| parser.add_argument('-vcfInfo', '--vcfInfo', type=str, help='The txt file of variants information, this file is named as prefix__variant_quality_location.txt', required=True) | |||
| parser.add_argument('-mendelianInfo', '--mendelianInfo', type=str, help='The merged mendelian information of all samples', required=True) | |||
| parser.add_argument('-sample', '--sample_name', type=str, help='which sample of quartet', required=True) | |||
| args = parser.parse_args() | |||
| vcfInfo = args.vcfInfo | |||
| mendelianInfo = args.mendelianInfo | |||
| sample_name = args.sample_name | |||
| #GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200331 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=1,Type=Flag,Description="1 for sister consistent, 0 for sister different"> | |||
| ##FORMAT=<ID=TRIO5,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL5 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=TRIO6,Number=1,Type=Flag,Description="1 for LCL7, LCL8 and LCL6 mendelian consistent, 0 for family violation"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Depth"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Alternative Depth"> | |||
| ##FORMAT=<ID=AF,Number=1,Type=Float,Description="Allele frequency"> | |||
| ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype quality"> | |||
| ##FORMAT=<ID=QD,Number=1,Type=Float,Description="Variant Confidence/Quality by Depth"> | |||
| ##FORMAT=<ID=MQ,Number=1,Type=Float,Description="Mapping quality"> | |||
| ##FORMAT=<ID=FS,Number=1,Type=Float,Description="Phred-scaled p-value using Fisher's exact test to detect strand bias"> | |||
| ##FORMAT=<ID=QUAL,Number=1,Type=Float,Description="variant quality"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # output file | |||
| file_name = sample_name + '_mendelian_vcfInfo.vcf' | |||
| outfile = open(file_name,'w') | |||
| outputcolumn = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t' + sample_name + '\n' | |||
| outfile.write(vcf_header) | |||
| outfile.write(outputcolumn) | |||
| # input files | |||
| vcf_info = pd.read_table(vcfInfo) | |||
| mendelian_info = pd.read_table(mendelianInfo) | |||
| merged_df = pd.merge(vcf_info, mendelian_info, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_df = merged_df.fillna('.') | |||
| # | |||
| def parse_INFO(info): | |||
| strings = info.strip().split(';') | |||
| keys = [] | |||
| values = [] | |||
| for i in strings: | |||
| kv = i.split('=') | |||
| if kv[0] == 'DB': | |||
| keys.append('DB') | |||
| values.append('1') | |||
| else: | |||
| keys.append(kv[0]) | |||
| values.append(kv[1]) | |||
| infoDict = dict(zip(keys, values)) | |||
| return infoDict | |||
| # | |||
| for row in merged_df.itertuples(): | |||
| if row[18] != '.': | |||
| # format | |||
| # GT:TWINS:TRIO5:TRIO6:DP:AF:GQ:QD:MQ:FS:QUAL | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = row[18] + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_y + '\t' + row.ALT_y + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL' + '\t' + FORMAT + '\n' | |||
| else: | |||
| rawGT = row[10].split(':') | |||
| FORMAT_x = row[10].split(':') | |||
| ALT = int(FORMAT_x[1].split(',')[1]) | |||
| if int(FORMAT_x[2]) != 0: | |||
| AF = round(ALT/int(FORMAT_x[2]),2) | |||
| else: | |||
| AF = '.' | |||
| INFO_x = parse_INFO(row.INFO_x) | |||
| if FORMAT_x[2] == '0': | |||
| INFO_x['QD'] = '.' | |||
| else: | |||
| pass | |||
| FORMAT = '.:.:.:.' + ':' + FORMAT_x[2] + ':' + str(ALT) + ':' + str(AF) + ':' + FORMAT_x[3] + ':' + INFO_x['QD'] + ':' + INFO_x['MQ'] + ':' + INFO_x['FS'] + ':' + str(row.QUAL_x) + ':' + rawGT[0] | |||
| # outline | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + row.ID_x + '\t' + row.REF_x + '\t' + row.ALT_x + '\t' + '.' + '\t' + '.' + '\t' + '.' + '\t' + 'GT:TWINS:TRIO5:TRIO6:DP:ALT:AF:GQ:QD:MQ:FS:QUAL:rawGT' + '\t' + FORMAT + '\n' | |||
| outfile.write(outline) | |||
+ 71
- 0
codescripts/merge_two_family.py
Прегледај датотеку
| @@ -0,0 +1,71 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to extract mendelian concordance information") | |||
| parser.add_argument('-LCL5', '--LCL5', type=str, help='LCL5 family info', required=True) | |||
| parser.add_argument('-LCL6', '--LCL6', type=str, help='LCL6 family info', required=True) | |||
| parser.add_argument('-family', '--family', type=str, help='family name', required=True) | |||
| args = parser.parse_args() | |||
| lcl5 = args.LCL5 | |||
| lcl6 = args.LCL6 | |||
| family = args.family | |||
| # output file | |||
| family_name = family + '.txt' | |||
| family_file = open(family_name,'w') | |||
| # input files | |||
| lcl5_dat = pd.read_table(lcl5) | |||
| lcl6_dat = pd.read_table(lcl6) | |||
| merged_df = pd.merge(lcl5_dat, lcl6_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| def alt_seq(alt, genotype): | |||
| if genotype == './.': | |||
| seq = './.' | |||
| elif genotype == '0/0': | |||
| seq = '0/0' | |||
| else: | |||
| alt = alt.split(',') | |||
| genotype = genotype.split('/') | |||
| if genotype[0] == '0': | |||
| allele2 = alt[int(genotype[1]) - 1] | |||
| seq = '0/' + allele2 | |||
| else: | |||
| allele1 = alt[int(genotype[0]) - 1] | |||
| allele2 = alt[int(genotype[1]) - 1] | |||
| seq = allele1 + '/' + allele2 | |||
| return seq | |||
| for row in merged_df.itertuples(): | |||
| # correction of multiallele | |||
| if pd.isnull(row.INFO_x) == True or pd.isnull(row.INFO_y) == True: | |||
| mendelian = '.' | |||
| else: | |||
| lcl5_seq = alt_seq(row.ALT_x, row.CHILD_x) | |||
| lcl6_seq = alt_seq(row.ALT_y, row.CHILD_y) | |||
| if lcl5_seq == lcl6_seq: | |||
| mendelian = '1' | |||
| else: | |||
| mendelian = '0' | |||
| if pd.isnull(row.INFO_x) == True: | |||
| mendelian = mendelian + ':.' | |||
| else: | |||
| mendelian = mendelian + ':' + row.INFO_x.split('=')[1] | |||
| if pd.isnull(row.INFO_y) == True: | |||
| mendelian = mendelian + ':.' | |||
| else: | |||
| mendelian = mendelian + ':' + row.INFO_y.split('=')[1] | |||
| outline = row._1 + '\t' + str(row.POS) + '\t' + mendelian + '\n' | |||
| family_file.write(outline) | |||
+ 161
- 0
codescripts/merge_two_family_with_genotype.py
Прегледај датотеку
| @@ -0,0 +1,161 @@ | |||
| from __future__ import division | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to extract mendelian concordance information") | |||
| parser.add_argument('-LCL5', '--LCL5', type=str, help='LCL5 family info', required=True) | |||
| parser.add_argument('-LCL6', '--LCL6', type=str, help='LCL6 family info', required=True) | |||
| parser.add_argument('-genotype', '--genotype', type=str, help='Genotype information of a set of four family members', required=True) | |||
| parser.add_argument('-family', '--family', type=str, help='family name', required=True) | |||
| args = parser.parse_args() | |||
| lcl5 = args.LCL5 | |||
| lcl6 = args.LCL6 | |||
| genotype = args.genotype | |||
| family = args.family | |||
| # output file | |||
| family_name = family + '.txt' | |||
| family_file = open(family_name,'w') | |||
| summary_name = family + '.summary.txt' | |||
| summary_file = open(summary_name,'w') | |||
| # input files | |||
| lcl5_dat = pd.read_table(lcl5) | |||
| lcl6_dat = pd.read_table(lcl6) | |||
| genotype_dat = pd.read_table(genotype) | |||
| merged_df = pd.merge(lcl5_dat, lcl6_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_genotype_df = pd.merge(merged_df, genotype_dat, how='outer', left_on=['#CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_genotype_df = merged_genotype_df.dropna() | |||
| merged_genotype_df_sub = merged_genotype_df.iloc[:,[0,1,23,24,29,30,31,32,7,17]] | |||
| merged_genotype_df_sub.columns = ['CHROM', 'POS', 'REF', 'ALT','LCL5','LCL6','LCL7','LCL8', 'TRIO5', 'TRIO6'] | |||
| indel_sister_same = 0 | |||
| indel_sister_diff = 0 | |||
| indel_family_all = 0 | |||
| indel_family_mendelian = 0 | |||
| snv_sister_same = 0 | |||
| snv_sister_diff = 0 | |||
| snv_family_all = 0 | |||
| snv_family_mendelian = 0 | |||
| for row in merged_genotype_df_sub.itertuples(): | |||
| # indel or snv | |||
| if ',' in row.ALT: | |||
| alt = row.ALT.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.ALT) | |||
| alt = alt_max | |||
| ref = row.REF | |||
| if len(ref) == 1 and alt == 1: | |||
| cate = 'SNV' | |||
| elif len(ref) > alt: | |||
| cate = 'INDEL' | |||
| elif alt > len(ref): | |||
| cate = 'INDEL' | |||
| elif len(ref) == alt: | |||
| cate = 'INDEL' | |||
| # sister | |||
| if row.LCL5 == row.LCL6: | |||
| if row.LCL5 == './.': | |||
| mendelian = 'noInfo' | |||
| sister_count = "no" | |||
| elif row.LCL5 == '0/0': | |||
| mendelian = 'Ref' | |||
| sister_count = "no" | |||
| else: | |||
| mendelian = '1' | |||
| sister_count = "yes_same" | |||
| else: | |||
| mendelian = '0' | |||
| if (row.LCL5 == './.' or row.LCL5 == '0/0') and (row.LCL6 == './.' or row.LCL6 == '0/0'): | |||
| sister_count = "no" | |||
| else: | |||
| sister_count = "yes_diff" | |||
| # family trio5 | |||
| if row.LCL5 == row.LCL7 == row.LCL8 == './.': | |||
| mendelian = mendelian + ':noInfo' | |||
| elif row.LCL5 == row.LCL7 == row.LCL8 == '0/0': | |||
| mendelian = mendelian + ':Ref' | |||
| elif pd.isnull(row.TRIO5) == True: | |||
| mendelian = mendelian + ':unVBT' | |||
| else: | |||
| mendelian = mendelian + ':' + row.TRIO5.split('=')[1] | |||
| # family trio6 | |||
| if row.LCL6 == row.LCL7 == row.LCL8 == './.': | |||
| mendelian = mendelian + ':noInfo' | |||
| elif row.LCL6 == row.LCL7 == row.LCL8 == '0/0': | |||
| mendelian = mendelian + ':Ref' | |||
| elif pd.isnull(row.TRIO6) == True: | |||
| mendelian = mendelian + ':unVBT' | |||
| else: | |||
| mendelian = mendelian + ':' + row.TRIO6.split('=')[1] | |||
| # not count into family | |||
| if (row.LCL5 == './.' or row.LCL5 == '0/0') and (row.LCL6 == './.' or row.LCL6 == '0/0') and (row.LCL7 == './.' or row.LCL7 == '0/0') and (row.LCL8 == './.' or row.LCL8 == '0/0'): | |||
| mendelian_count = "no" | |||
| else: | |||
| mendelian_count = "yes" | |||
| outline = str(row.CHROM) + '\t' + str(row.POS) + '\t' + str(row.REF) + '\t' + str(row.ALT) + '\t' + str(cate) + '\t' + str(row.LCL5) + '\t' + str(row.LCL6) + '\t' + str(row.LCL7) + '\t' + str(row.LCL8) + '\t' + str(row.TRIO5) + '\t' + str(row.TRIO6) + '\t' + str(mendelian) + '\t' + str(mendelian_count) + '\t' + str(sister_count) + '\n' | |||
| family_file.write(outline) | |||
| if cate == 'SNV': | |||
| if sister_count == 'yes_same': | |||
| snv_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| snv_sister_diff += 1 | |||
| else: | |||
| pass | |||
| if mendelian_count == 'yes': | |||
| snv_family_all += 1 | |||
| else: | |||
| pass | |||
| if mendelian == '1:1:1': | |||
| snv_family_mendelian += 1 | |||
| elif mendelian == 'Ref:1:1': | |||
| snv_family_mendelian += 1 | |||
| else: | |||
| pass | |||
| elif cate == 'INDEL': | |||
| if sister_count == 'yes_same': | |||
| indel_sister_same += 1 | |||
| elif sister_count == 'yes_diff': | |||
| indel_sister_diff += 1 | |||
| else: | |||
| pass | |||
| if mendelian_count == 'yes': | |||
| indel_family_all += 1 | |||
| else: | |||
| pass | |||
| if mendelian == '1:1:1': | |||
| indel_family_mendelian += 1 | |||
| elif mendelian == 'Ref:1:1': | |||
| indel_family_mendelian += 1 | |||
| else: | |||
| pass | |||
| snv_sister = snv_sister_same/(snv_sister_same + snv_sister_diff) | |||
| indel_sister = indel_sister_same/(indel_sister_same + indel_sister_diff) | |||
| snv_quartet = snv_family_mendelian/snv_family_all | |||
| indel_quartet = indel_family_mendelian/indel_family_all | |||
| outcolumn = 'Family\tReproducibility_D5_D6\tMendelian_Concordance_Quartet\n' | |||
| indel_outResult = family + '.INDEL' + '\t' + str(indel_sister) + '\t' + str(indel_quartet) + '\n' | |||
| snv_outResult = family + '.SNV' + '\t' + str(snv_sister) + '\t' + str(snv_quartet) + '\n' | |||
| summary_file.write(outcolumn) | |||
| summary_file.write(indel_outResult) | |||
| summary_file.write(snv_outResult) | |||
+ 109
- 0
codescripts/oneClass.py
Прегледај датотеку
| @@ -0,0 +1,109 @@ | |||
| # import modules | |||
| import numpy as np | |||
| import pandas as pd | |||
| from sklearn import svm | |||
| from sklearn import preprocessing | |||
| import sys, argparse, os | |||
| from vcf2bed import position_to_bed,padding_region | |||
| parser = argparse.ArgumentParser(description="this script is to preform one calss svm on each chromosome") | |||
| parser.add_argument('-train', '--trainDataset', type=str, help='training dataset generated from extracting vcf information part, with mutaitons supported by callsets', required=True) | |||
| parser.add_argument('-test', '--testDataset', type=str, help='testing dataset generated from extracting vcf information part, with mutaitons not called by all callsets', required=True) | |||
| parser.add_argument('-name', '--sampleName', type=str, help='sample name for output file name', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| train_input = args.trainDataset | |||
| test_input = args.testDataset | |||
| sample_name = args.sampleName | |||
| # default columns, which will be included in the included in the calssifier | |||
| chromosome = ['chr1','chr2','chr3','chr4','chr5','chr6','chr7','chr8','chr9','chr10','chr11','chr12','chr13','chr14','chr15' ,'chr16','chr17','chr18','chr19','chr20','chr21','chr22','chrX','chrY'] | |||
| feature_heter_cols = ['AltDP','BaseQRankSum','DB','DP','FS','GQ','MQ','MQRankSum','QD','ReadPosRankSum','RefDP','SOR','af'] | |||
| feature_homo_cols = ['AltDP','DB','DP','FS','GQ','MQ','QD','RefDP','SOR','af'] | |||
| # import datasets sepearate the records with or without BaseQRankSum annotation, etc. | |||
| def load_dat(dat_file_name): | |||
| dat = pd.read_table(dat_file_name) | |||
| dat['DB'] = dat['DB'].fillna(0) | |||
| dat = dat[dat['DP'] != 0] | |||
| dat['af'] = dat['AltDP']/(dat['AltDP'] + dat['RefDP']) | |||
| homo_rows = dat[dat['BaseQRankSum'].isnull()] | |||
| heter_rows = dat[dat['BaseQRankSum'].notnull()] | |||
| return homo_rows,heter_rows | |||
| train_homo,train_heter = load_dat(train_input) | |||
| test_homo,test_heter = load_dat(test_input) | |||
| clf = svm.OneClassSVM(nu=0.05,kernel='rbf', gamma='auto_deprecated',cache_size=500) | |||
| def prepare_dat(train_dat,test_dat,feature_cols,chromo): | |||
| chr_train = train_dat[train_dat['chromo'] == chromo] | |||
| chr_test = test_dat[test_dat['chromo'] == chromo] | |||
| train_dat = chr_train.loc[:,feature_cols] | |||
| test_dat = chr_test.loc[:,feature_cols] | |||
| train_dat_scaled = preprocessing.scale(train_dat) | |||
| test_dat_scaled = preprocessing.scale(test_dat) | |||
| return chr_test,train_dat_scaled,test_dat_scaled | |||
| def oneclass(X_train,X_test,chr_test): | |||
| clf.fit(X_train) | |||
| y_pred_test = clf.predict(X_test) | |||
| test_true_dat = chr_test[y_pred_test == 1] | |||
| test_false_dat = chr_test[y_pred_test == -1] | |||
| return test_true_dat,test_false_dat | |||
| predicted_true = pd.DataFrame(columns=train_homo.columns) | |||
| predicted_false = pd.DataFrame(columns=train_homo.columns) | |||
| for chromo in chromosome: | |||
| # homo datasets | |||
| chr_test_homo,X_train_homo,X_test_homo = prepare_dat(train_homo,test_homo,feature_homo_cols,chromo) | |||
| test_true_homo,test_false_homo = oneclass(X_train_homo,X_test_homo,chr_test_homo) | |||
| predicted_true = predicted_true.append(test_true_homo) | |||
| predicted_false = predicted_false.append(test_false_homo) | |||
| # heter datasets | |||
| chr_test_heter,X_train_heter,X_test_heter = prepare_dat(train_heter,test_heter,feature_heter_cols,chromo) | |||
| test_true_heter,test_false_heter = oneclass(X_train_heter,X_test_heter,chr_test_heter) | |||
| predicted_true = predicted_true.append(test_true_heter) | |||
| predicted_false = predicted_false.append(test_false_heter) | |||
| predicted_true_filename = sample_name + '_predicted_true.txt' | |||
| predicted_false_filename = sample_name + '_predicted_false.txt' | |||
| predicted_true.to_csv(predicted_true_filename,sep='\t',index=False) | |||
| predicted_false.to_csv(predicted_false_filename,sep='\t',index=False) | |||
| # output the bed file and padding bed region 50bp | |||
| predicted_true_bed_filename = sample_name + '_predicted_true.bed' | |||
| predicted_false_bed_filename = sample_name + '_predicted_false.bed' | |||
| padding_filename = sample_name + '_padding.bed' | |||
| predicted_true_bed = open(predicted_true_bed_filename,'w') | |||
| predicted_false_bed = open(predicted_false_bed_filename,'w') | |||
| padding = open(padding_filename,'w') | |||
| # | |||
| for index,row in predicted_false.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_false_bed.write(outline_pos) | |||
| chromo,pad_pos1,pad_pos2,pad_pos3,pad_pos4 = padding_region(chromo,pos1,pos2,50) | |||
| outline_pad_1 = chromo + '\t' + str(pad_pos1) + '\t' + str(pad_pos2) + '\n' | |||
| outline_pad_2 = chromo + '\t' + str(pad_pos3) + '\t' + str(pad_pos4) + '\n' | |||
| padding.write(outline_pad_1) | |||
| padding.write(outline_pad_2) | |||
| for index,row in predicted_true.iterrows(): | |||
| chromo,pos1,pos2 = position_to_bed(row['chromo'],row['pos'],row['ref'],row['alt']) | |||
| outline_pos = chromo + '\t' + str(pos1) + '\t' + str(pos2) + '\n' | |||
| predicted_true_bed.write(outline_pos) | |||
+ 101
- 0
codescripts/post-alignment.py
Прегледај датотеку
| @@ -0,0 +1,101 @@ | |||
| import json | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import statistics | |||
| parser = argparse.ArgumentParser(description="This script is to summary information for pre-alignment QC") | |||
| parser.add_argument('-general', '--general_stat', type=str, help='multiqc_general_stats.txt', required=True) | |||
| parser.add_argument('-is', '--is_metrics', type=str, help='_is_metrics.txt', required=True) | |||
| parser.add_argument('-wgsmetrics', '--WgsMetricsAlgo', type=str, help='deduped_WgsMetricsAlgo', required=True) | |||
| parser.add_argument('-qualityyield', '--QualityYield', type=str, help='deduped_QualityYield', required=True) | |||
| parser.add_argument('-aln', '--aln_metrics', type=str, help='aln_metrics.txt', required=True) | |||
| args = parser.parse_args() | |||
| general_file = args.general_stat | |||
| is_file = args.is_metrics | |||
| wgsmetrics_file = args.wgsmetrics | |||
| qualityyield_file = args.qualityyield | |||
| aln_file = args.aln_metrics | |||
| ##### Table | |||
| ## general stat: % GC | |||
| dat = pd.read_table(general_file) | |||
| qualimap = dat.loc[:, dat.columns.str.startswith('QualiMap')] | |||
| qualimap.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| qualimap_stat = qualimap.dropna() | |||
| part1 = fastqc_stat.loc[:,['Sample', 'FastQC_mqc-generalstats-fastqc-percent_duplicates','FastQC_mqc-generalstats-fastqc-total_sequences']] | |||
| ## is_metrics: median insert size | |||
| ## deduped_WgsMetricsAlgo: 1x, 5x, 10x, 30x, median coverage | |||
| with open(html_file) as file: | |||
| origDict = json.load(file) | |||
| newdict = {(k1, k2):v2 for k1,v1 in origDict.items() \ | |||
| for k2,v2 in origDict[k1].items()} | |||
| df = pd.DataFrame([newdict[i] for i in sorted(newdict)], | |||
| index=pd.MultiIndex.from_tuples([i for i in sorted(newdict.keys())])) | |||
| gc = [] | |||
| at = [] | |||
| for i in part1['Sample']: | |||
| sub_df = df.loc[i,:] | |||
| gc.append(statistics.mean(sub_df['g']/sub_df['c'])) | |||
| at.append(statistics.mean(sub_df['a']/sub_df['t'])) | |||
| ## fastq_screen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| del fastqscreen['ERCC percentage'] | |||
| del fastqscreen['Phix percentage'] | |||
| ### merge all information | |||
| part1.insert(loc=3, column='G/C ratio', value=gc) | |||
| part1.insert(loc=4, column='A/T ratio', value=at) | |||
| part1.reset_index(drop=True, inplace=True) | |||
| fastqscreen.reset_index(drop=True, inplace=True) | |||
| df = pd.concat([part1, fastqscreen], axis=1) | |||
| df = df.append(df.mean(axis=0),ignore_index=True) | |||
| df = df.fillna('Batch average value') | |||
| df.columns = ['Sample','Total sequences (million)','% Dup','G/C ratio','A/T ratio','% Human','% EColi','% Adapter' , '% Vector','% rRNA' , '% Virus','% Yeast' ,'% Mitoch' ,'% No hits'] | |||
| df.to_csv('per-alignment_table_summary.txt',sep='\t',index=False) | |||
| ##### Picture | |||
| ## cumulative genome coverage | |||
| with open(json_file) as file: | |||
| all_dat = json.load(file) | |||
| genome_coverage_json = all_dat['report_plot_data']['qualimap_genome_fraction']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(genome_coverage_json) | |||
| genome_coverage = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| genome_coverage[dat.loc[i,'name']] = one_sample[1] | |||
| genome_coverage = genome_coverage.transpose() | |||
| genome_coverage['Sample'] = genome_coverage.index | |||
| genome_coverage.to_csv('post-alignment_genome_coverage.txt',sep='\t',index=False) | |||
| ## insert size histogram | |||
| insert_size_json = all_dat['report_plot_data']['qualimap_insert_size']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(insert_size_json) | |||
| insert_size = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| insert_size[dat.loc[i,'name']] = one_sample[1] | |||
| insert_size = insert_size.transpose() | |||
| insert_size['Sample'] = insert_size.index | |||
| insert_size.to_csv('post-alignment_insert_size.txt',sep='\t',index=False) | |||
| ## GC content distribution | |||
| gc_content_json = all_dat['report_plot_data']['qualimap_gc_content']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(gc_content_json) | |||
| gc_content = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| gc_content[dat.loc[i,'name']] = one_sample[1] | |||
| gc_content = gc_content.transpose() | |||
| gc_content['Sample'] = gc_content.index | |||
| gc_content.to_csv('post-alignment_gc_content.txt',sep='\t',index=False) | |||
+ 132
- 0
codescripts/pre_alignment.py
Прегледај датотеку
| @@ -0,0 +1,132 @@ | |||
| import json | |||
| import pandas as pd | |||
| import sys, argparse, os | |||
| import statistics | |||
| parser = argparse.ArgumentParser(description="This script is to summary information for pre-alignment QC") | |||
| parser.add_argument('-general', '--general_stat', type=str, help='multiqc_general_stats.txt', required=True) | |||
| parser.add_argument('-html', '--html', type=str, help='multiqc_report.html', required=True) | |||
| parser.add_argument('-fastqscreen', '--fastqscreen', type=str, help='multiqc_fastq_screen.txt', required=True) | |||
| parser.add_argument('-json', '--json', type=str, help='multiqc_happy_data.json', required=True) | |||
| args = parser.parse_args() | |||
| general_file = args.general_stat | |||
| html_file = args.html | |||
| fastqscreen_file = args.fastqscreen | |||
| json_file = args.json | |||
| ##### Table | |||
| ## general stat: 1. Total sequences; 2. %Dup | |||
| dat = pd.read_table(general_file) | |||
| fastqc = dat.loc[:, dat.columns.str.startswith('FastQC')] | |||
| fastqc.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_stat = fastqc.dropna() | |||
| part1 = fastqc_stat.loc[:,['Sample', 'FastQC_mqc-generalstats-fastqc-percent_duplicates','FastQC_mqc-generalstats-fastqc-total_sequences']] | |||
| ## report html: 1. G/C ratio; 2. A/T ratio | |||
| ## cat multiqc_report.html | grep 'fastqc_seq_content_data = ' | sed s'/fastqc_seq_content_data\ =\ //g' | sed 's/^[ \t]*//g' | sed s'/;//g' > fastqc_sequence_content.json | |||
| with open(html_file) as file: | |||
| origDict = json.load(file) | |||
| newdict = {(k1, k2):v2 for k1,v1 in origDict.items() \ | |||
| for k2,v2 in origDict[k1].items()} | |||
| df = pd.DataFrame([newdict[i] for i in sorted(newdict)], | |||
| index=pd.MultiIndex.from_tuples([i for i in sorted(newdict.keys())])) | |||
| gc = [] | |||
| at = [] | |||
| for i in part1['Sample']: | |||
| sub_df = df.loc[i,:] | |||
| gc.append(statistics.mean(sub_df['g']/sub_df['c'])) | |||
| at.append(statistics.mean(sub_df['a']/sub_df['t'])) | |||
| ## fastq_screen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| del fastqscreen['ERCC percentage'] | |||
| del fastqscreen['Phix percentage'] | |||
| ### merge all information | |||
| part1.insert(loc=3, column='G/C ratio', value=gc) | |||
| part1.insert(loc=4, column='A/T ratio', value=at) | |||
| part1.reset_index(drop=True, inplace=True) | |||
| fastqscreen.reset_index(drop=True, inplace=True) | |||
| df = pd.concat([part1, fastqscreen], axis=1) | |||
| df = df.append(df.mean(axis=0),ignore_index=True) | |||
| df = df.fillna('Batch average value') | |||
| df.columns = ['Sample','Total sequences (million)','% Dup','G/C ratio','A/T ratio','% Human','% EColi','% Adapter' , '% Vector','% rRNA' , '% Virus','% Yeast' ,'% Mitoch' ,'% No hits'] | |||
| df.to_csv('per-alignment_table_summary.txt',sep='\t',index=False) | |||
| ##### Picture | |||
| ## mean quality scores | |||
| with open(json_file) as file: | |||
| all_dat = json.load(file) | |||
| mean_quality_json = all_dat['report_plot_data']['fastqc_per_base_sequence_quality_plot']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(mean_quality_json) | |||
| mean_quality = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| mean_quality[dat.loc[i,'name']] = one_sample[1] | |||
| mean_quality = mean_quality.transpose() | |||
| mean_quality['Sample'] = mean_quality.index | |||
| mean_quality.to_csv('pre-alignment_mean_quality.txt',sep='\t',index=False) | |||
| ## per sequence GC content | |||
| gc_content_json = all_dat['report_plot_data']['fastqc_per_sequence_gc_content_plot']['datasets'][0] | |||
| dat =pd.DataFrame.from_records(gc_content_json) | |||
| gc_content = pd.DataFrame(index=pd.DataFrame(dat.loc[0,'data'])[0]) | |||
| for i in range(dat.shape[0]): | |||
| one_sample = pd.DataFrame(dat.loc[i,'data']) | |||
| one_sample.index = one_sample[0] | |||
| gc_content[dat.loc[i,'name']] = one_sample[1] | |||
| gc_content = gc_content.transpose() | |||
| gc_content['Sample'] = gc_content.index | |||
| gc_content.to_csv('pre-alignment_gc_content.txt',sep='\t',index=False) | |||
| # fastqc and qualimap | |||
| dat = pd.read_table(fastqc_qualimap_file) | |||
| fastqc = dat.loc[:, dat.columns.str.startswith('FastQC')] | |||
| fastqc.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_stat = fastqc.dropna() | |||
| # qulimap | |||
| qualimap = dat.loc[:, dat.columns.str.startswith('QualiMap')] | |||
| qualimap.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| qualimap_stat = qualimap.dropna() | |||
| # fastqc | |||
| dat = pd.read_table(fastqc_file) | |||
| fastqc_module = dat.loc[:, "per_base_sequence_quality":"kmer_content"] | |||
| fastqc_module.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| fastqc_all = pd.merge(fastqc_stat,fastqc_module, how='outer', left_on=['Sample'], right_on = ['Sample']) | |||
| # fastqscreen | |||
| dat = pd.read_table(fastqscreen_file) | |||
| fastqscreen = dat.loc[:, dat.columns.str.endswith('percentage')] | |||
| dat['Sample'] = [i.replace('_screen','') for i in dat['Sample']] | |||
| fastqscreen.insert(loc=0, column='Sample', value=dat['Sample']) | |||
| # benchmark | |||
| with open(hap_file) as hap_json: | |||
| happy = json.load(hap_json) | |||
| dat =pd.DataFrame.from_records(happy) | |||
| dat = dat.loc[:, dat.columns.str.endswith('ALL')] | |||
| dat_transposed = dat.T | |||
| benchmark = dat_transposed.loc[:,['sample_id','METRIC.Precision','METRIC.Recall']] | |||
| benchmark['sample_id'] = benchmark.index | |||
| benchmark.columns = ['Sample','Precision','Recall'] | |||
| #output | |||
| fastqc_all.to_csv('fastqc.final.result.txt',sep="\t",index=0) | |||
| fastqscreen.to_csv('fastqscreen.final.result.txt',sep="\t",index=0) | |||
| qualimap_stat.to_csv('qualimap.final.result.txt',sep="\t",index=0) | |||
| benchmark.to_csv('benchmark.final.result.txt',sep="\t",index=0) | |||
+ 144
- 0
codescripts/reformVCF.py
Прегледај датотеку
| @@ -0,0 +1,144 @@ | |||
| # import modules | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| parser = argparse.ArgumentParser(description="This script is to split samples in VCF files and rewrite to the right style") | |||
| parser.add_argument('-vcf', '--familyVCF', type=str, help='VCF with sister and mendelian infomation', required=True) | |||
| parser.add_argument('-name', '--familyName', type=str, help='Family name of the VCF file', required=True) | |||
| args = parser.parse_args() | |||
| # Rename input: | |||
| inputFile = args.familyVCF | |||
| family_name = args.familyName | |||
| # output filename | |||
| LCL5_name = family_name + '.LCL5.vcf' | |||
| LCL5file = open(LCL5_name,'w') | |||
| LCL6_name = family_name + '.LCL6.vcf' | |||
| LCL6file = open(LCL6_name,'w') | |||
| LCL7_name = family_name + '.LCL7.vcf' | |||
| LCL7file = open(LCL7_name,'w') | |||
| LCL8_name = family_name + '.LCL8.vcf' | |||
| LCL8file = open(LCL8_name,'w') | |||
| family_filename = family_name + '.vcf' | |||
| familyfile = open(family_filename,'w') | |||
| # default columns, which will be included in the included in the calssifier | |||
| vcfheader = '''##fileformat=VCFv4.2 | |||
| ##FILTER=<ID=PASS,Description="the same genotype between twin sister and mendelian consistent in 578 and 678"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=TWINS,Number=0,Type=Flag,Description="0 for sister consistent, 1 for sister inconsistent"> | |||
| ##FORMAT=<ID=TRIO5,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##FORMAT=<ID=TRIO6,Number=0,Type=Flag,Description="0 for trio consistent, 1 for trio inconsistent"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # write VCF | |||
| LCL5colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL5'+'\n' | |||
| LCL5file.write(vcfheader) | |||
| LCL5file.write(LCL5colname) | |||
| LCL6colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL6'+'\n' | |||
| LCL6file.write(vcfheader) | |||
| LCL6file.write(LCL6colname) | |||
| LCL7colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL7'+'\n' | |||
| LCL7file.write(vcfheader) | |||
| LCL7file.write(LCL7colname) | |||
| LCL8colname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+family_name+'_LCL8'+'\n' | |||
| LCL8file.write(vcfheader) | |||
| LCL8file.write(LCL8colname) | |||
| familycolname = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\t'+'LCL5\t'+'LCL6\t'+'LCL7\t'+'LCL8'+'\n' | |||
| familyfile.write(vcfheader) | |||
| familyfile.write(familycolname) | |||
| # reform VCF | |||
| def process(oneLine): | |||
| line = oneLine.rstrip() | |||
| strings = line.strip().split('\t') | |||
| # replace . | |||
| # LCL6 uniq | |||
| if strings[11] == '.': | |||
| strings[11] = '0/0' | |||
| strings[9] = strings[12] | |||
| strings[10] = strings[13] | |||
| else: | |||
| pass | |||
| # LCL5 uniq | |||
| if strings[14] == '.': | |||
| strings[14] = '0/0' | |||
| strings[12] = strings[9] | |||
| strings[13] = strings[10] | |||
| else: | |||
| pass | |||
| # sister | |||
| if strings[11] == strings[14]: | |||
| add_format = ":1" | |||
| else: | |||
| add_format = ":0" | |||
| # trioLCL5 | |||
| if strings[15] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # trioLCL6 | |||
| if strings[7] == 'MD=1': | |||
| add_format = add_format + ":1" | |||
| else: | |||
| add_format = add_format + ":0" | |||
| # filter | |||
| if (strings[11] == strings[14]) and (strings[15] == 'MD=1') and (strings[7] == 'MD=1'): | |||
| strings[6] = 'PASS' | |||
| else: | |||
| strings[6] = '.' | |||
| # output LCL5 | |||
| LCL5outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format + '\n' | |||
| LCL5file.write(LCL5outLine) | |||
| # output LCL6 | |||
| LCL6outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[11] + add_format + '\n' | |||
| LCL6file.write(LCL6outLine) | |||
| # output LCL7 | |||
| LCL7outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[10] + add_format + '\n' | |||
| LCL7file.write(LCL7outLine) | |||
| # output LCL8 | |||
| LCL8outLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+'.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[9] + add_format + '\n' | |||
| LCL8file.write(LCL8outLine) | |||
| # output family | |||
| familyoutLine = strings[0]+'\t'+strings[1]+'\t'+strings[2]+'\t'+strings[3]+'\t'+strings[4]+'\t'+ '.'+'\t'+strings[6]+'\t'+ '.' +'\t'+ 'GT:TWINS:TRIO5:TRIO6' + '\t' + strings[14] + add_format +'\t' + strings[11] + add_format + '\t' + strings[10] + add_format +'\t' + strings[9] + add_format + '\n' | |||
| familyfile.write(familyoutLine) | |||
| for line in fileinput.input(inputFile): | |||
| m = re.match('^\#',line) | |||
| if m is not None: | |||
| pass | |||
| else: | |||
| process(line) | |||
+ 227
- 0
codescripts/replicates_consensus.py
Прегледај датотеку
| @@ -0,0 +1,227 @@ | |||
| from __future__ import division | |||
| from glob import glob | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-prefix', '--prefix', type=str, help='prefix of output file', required=True) | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| prefix = args.prefix | |||
| vcf = args.vcf | |||
| # input files | |||
| vcf_dat = pd.read_table(vcf) | |||
| # all info | |||
| all_file_name = prefix + "_all_summary.txt" | |||
| all_sample_outfile = open(all_file_name,'w') | |||
| all_info_col = 'CHROM\tPOS\tREF\tALT\tLCL5_consensus_calls\tLCL5_detect_number\tLCL5_same_diff\tLCL6_consensus_calls\tLCL6_detect_number\tLCl6_same_diff\tLCL7_consensus_calls\tLCL7_detect_number\tLCL7_same_diff\tLCL8_consensus_calls\tLCL8_detect_number\tLCL8_same_diff\n' | |||
| all_sample_outfile.write(all_info_col) | |||
| # filtered info | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200501 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| consensus_file_name = prefix + "_consensus.vcf" | |||
| consensus_outfile = open(consensus_file_name,'w') | |||
| consensus_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5_consensus_call\tLCL6_consensus_call\tLCL7_consensus_call\tLCL8_consensus_call\n' | |||
| consensus_outfile.write(vcf_header) | |||
| consensus_outfile.write(consensus_col) | |||
| # function | |||
| def decide_by_rep(vcf_list): | |||
| consensus_rep = '' | |||
| gt = [x.split(':')[0] for x in vcf_list] | |||
| gt_num_dict = Counter(gt) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if freq_gt >= 2: | |||
| consensus_rep = candidate_gt | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| return consensus_rep | |||
| def consensus_call(vcf_info_list): | |||
| consensus_call = '.' | |||
| detect_number = '.' | |||
| same_diff = '.' | |||
| # pcr | |||
| SEQ2000 = decide_by_rep(vcf_info_list[0:3]) | |||
| XTen_ARD = decide_by_rep(vcf_info_list[18:21]) | |||
| XTen_NVG = decide_by_rep(vcf_info_list[21:24]) | |||
| XTen_WUX = decide_by_rep(vcf_info_list[24:27]) | |||
| Nova_WUX = decide_by_rep(vcf_info_list[15:18]) | |||
| pcr_sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX,Nova_WUX] | |||
| pcr_sequence_dict = Counter(pcr_sequence_site) | |||
| pcr_highest_sequence = pcr_sequence_dict.most_common(1) | |||
| pcr_candidate_sequence = pcr_highest_sequence[0][0] | |||
| pcr_freq_sequence = pcr_highest_sequence[0][1] | |||
| if pcr_freq_sequence > 3: | |||
| pcr_consensus = pcr_candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| T7_WGE = decide_by_rep(vcf_info_list[3:6]) | |||
| Nova_ARD_1 = decide_by_rep(vcf_info_list[6:9]) | |||
| Nova_ARD_2 = decide_by_rep(vcf_info_list[9:12]) | |||
| Nova_BRG = decide_by_rep(vcf_info_list[12:15]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 2: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| gt = [x.replace('./.','.') for x in gt] | |||
| detected_num = 27 - gt.count('.') | |||
| gt_remain = [e for e in gt if e not in {'.'}] | |||
| gt_set = set(gt_remain) | |||
| if len(gt_set) == 1: | |||
| same_diff = 'same' | |||
| else: | |||
| same_diff = 'diff' | |||
| tag = ['inconGT','inconSequenceSite'] | |||
| if (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag): | |||
| consensus_call = pcr_consensus | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'notAgree' | |||
| else: | |||
| consensus_call = 'notConsensus' | |||
| return consensus_call, detected_num, same_diff | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| elif ((pcr_consensus == './.') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != './.')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| elif ((pcr_consensus != './.') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == './.')): | |||
| consensus_call = 'pcr-speicifc' | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = '0/0' | |||
| else: | |||
| consensus_call = 'filtered' | |||
| for row in vcf_dat.itertuples(): | |||
| # length | |||
| #alt | |||
| if ',' in row.ALT: | |||
| alt = row.ALT.split(',') | |||
| alt_len = [len(i) for i in alt] | |||
| alt_max = max(alt_len) | |||
| else: | |||
| alt_max = len(row.ALT) | |||
| #ref | |||
| ref_len = len(row.REF) | |||
| if (alt_max > 50) or (ref_len > 50): | |||
| pass | |||
| else: | |||
| # consensus | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl6_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL6_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL6_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL6_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL6_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL6_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL6_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_3_20170216] | |||
| lcl7_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL7_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL7_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL7_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL7_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL7_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL7_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_3_20170216] | |||
| lcl8_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL8_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL8_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL8_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL8_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL8_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL8_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_3_20170216] | |||
| # LCL5 | |||
| LCL5_consensus_call, LCL5_detected_num, LCL5_same_diff = consensus_call(lcl5_list) | |||
| # LCL6 | |||
| LCL6_consensus_call, LCL6_detected_num, LCL6_same_diff = consensus_call(lcl6_list) | |||
| # LCL7 | |||
| LCL7_consensus_call, LCL7_detected_num, LCL7_same_diff = consensus_call(lcl7_list) | |||
| # LCL8 | |||
| LCL8_consensus_call, LCL8_detected_num, LCL8_same_diff = consensus_call(lcl8_list) | |||
| # all data | |||
| all_output = row._1 + '\t' + str(row.POS) + '\t' + row.REF + '\t' + row.ALT + '\t' + LCL5_consensus_call + '\t' + str(LCL5_detected_num) + '\t' + LCL5_same_diff + '\t' +\ | |||
| LCL6_consensus_call + '\t' + str(LCL6_detected_num) + '\t' + LCL6_same_diff + '\t' +\ | |||
| LCL7_consensus_call + '\t' + str(LCL7_detected_num) + '\t' + LCL7_same_diff + '\t' +\ | |||
| LCL8_consensus_call + '\t' + str(LCL8_detected_num) + '\t' + LCL8_same_diff + '\n' | |||
| all_sample_outfile.write(all_output) | |||
| #consensus vcf | |||
| one_position = [LCL5_consensus_call,LCL6_consensus_call,LCL7_consensus_call,LCL8_consensus_call] | |||
| if ('notConsensus' in one_position) or (((len(set(one_position)) == 1) and ('./.' in set(one_position))) or ((len(set(one_position)) == 1) and ('0/0' in set(one_position))) or ((len(set(one_position)) == 2) and ('0/0' in set(one_position) and ('./.' in set(one_position))))): | |||
| pass | |||
| else: | |||
| consensus_output = row._1 + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + row.ALT + '\t' + '.' + '\t' + '.' + '\t' +'.' + '\t' + 'GT' + '\t' + LCL5_consensus_call + '\t' + LCL6_consensus_call + '\t' + LCL7_consensus_call + '\t' + LCL8_consensus_call +'\n' | |||
| consensus_outfile.write(consensus_output) | |||
+ 36
- 0
codescripts/vcf2bed.py
Прегледај датотеку
| @@ -0,0 +1,36 @@ | |||
| import re | |||
| def position_to_bed(chromo,pos,ref,alt): | |||
| # snv | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF | |||
| if len(ref) == 1 and len(alt) == 1: | |||
| StartPos = int(pos) -1 | |||
| EndPos = int(pos) | |||
| # deletions | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (reference length - alternate length) | |||
| elif len(ref) > 1 and len(alt) == 1: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(ref) - 1) | |||
| #insertions | |||
| # For insertions: | |||
| # Start cooridinate BED = start coordinate VCF - 1 | |||
| # End cooridinate BED = start coordinate VCF + (alternate length - reference length) | |||
| else: | |||
| StartPos = int(pos) - 1 | |||
| EndPos = int(pos) + (len(alt) - 1) | |||
| return chromo,StartPos,EndPos | |||
| def padding_region(chromo,pos1,pos2,padding): | |||
| StartPos1 = pos1 - padding | |||
| EndPos1 = pos1 | |||
| StartPos2 = pos2 | |||
| EndPos2 = pos2 + padding | |||
| return chromo,StartPos1,EndPos1,StartPos2,EndPos2 | |||
+ 295
- 0
codescripts/voted_by_vcfinfo_mendelianinfo.py
Прегледај датотеку
| @@ -0,0 +1,295 @@ | |||
| from __future__ import division | |||
| from glob import glob | |||
| import sys, argparse, os | |||
| import fileinput | |||
| import re | |||
| import pandas as pd | |||
| from operator import itemgetter | |||
| from collections import Counter | |||
| from itertools import islice | |||
| from numpy import * | |||
| import statistics | |||
| # input arguments | |||
| parser = argparse.ArgumentParser(description="this script is to merge mendelian and vcfinfo, and extract high_confidence_calls") | |||
| parser.add_argument('-folder', '--folder', type=str, help='directory that holds all the mendelian info', required=True) | |||
| parser.add_argument('-vcf', '--vcf', type=str, help='merged multiple sample vcf', required=True) | |||
| args = parser.parse_args() | |||
| folder = args.folder | |||
| vcf = args.vcf | |||
| # input files | |||
| folder = folder + '/*.txt' | |||
| filenames = glob(folder) | |||
| dataframes = [] | |||
| for filename in filenames: | |||
| dataframes.append(pd.read_table(filename,header=None)) | |||
| dfs = [df.set_index([0, 1]) for df in dataframes] | |||
| merged_mendelian = pd.concat(dfs, axis=1).reset_index() | |||
| family_name = [i.split('/')[-1].replace('.txt','') for i in filenames] | |||
| columns = ['CHROM','POS'] + family_name | |||
| merged_mendelian.columns = columns | |||
| vcf_dat = pd.read_table(vcf) | |||
| merged_df = pd.merge(merged_mendelian, vcf_dat, how='outer', left_on=['CHROM','POS'], right_on = ['#CHROM','POS']) | |||
| merged_df = merged_df.fillna('nan') | |||
| vcf_header = '''##fileformat=VCFv4.2 | |||
| ##fileDate=20200501 | |||
| ##source=high_confidence_calls_intergration(choppy app) | |||
| ##reference=GRCh38.d1.vd1 | |||
| ##INFO=<ID=VOTED,Number=1,Type=Integer,Description="Number mendelian consisitent votes"> | |||
| ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> | |||
| ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Sum depth of all samples"> | |||
| ##FORMAT=<ID=ALT,Number=1,Type=Integer,Description="Sum alternative depth of all samples"> | |||
| ##contig=<ID=chr1,length=248956422> | |||
| ##contig=<ID=chr2,length=242193529> | |||
| ##contig=<ID=chr3,length=198295559> | |||
| ##contig=<ID=chr4,length=190214555> | |||
| ##contig=<ID=chr5,length=181538259> | |||
| ##contig=<ID=chr6,length=170805979> | |||
| ##contig=<ID=chr7,length=159345973> | |||
| ##contig=<ID=chr8,length=145138636> | |||
| ##contig=<ID=chr9,length=138394717> | |||
| ##contig=<ID=chr10,length=133797422> | |||
| ##contig=<ID=chr11,length=135086622> | |||
| ##contig=<ID=chr12,length=133275309> | |||
| ##contig=<ID=chr13,length=114364328> | |||
| ##contig=<ID=chr14,length=107043718> | |||
| ##contig=<ID=chr15,length=101991189> | |||
| ##contig=<ID=chr16,length=90338345> | |||
| ##contig=<ID=chr17,length=83257441> | |||
| ##contig=<ID=chr18,length=80373285> | |||
| ##contig=<ID=chr19,length=58617616> | |||
| ##contig=<ID=chr20,length=64444167> | |||
| ##contig=<ID=chr21,length=46709983> | |||
| ##contig=<ID=chr22,length=50818468> | |||
| ##contig=<ID=chrX,length=156040895> | |||
| ''' | |||
| # output files | |||
| benchmark_LCL5 = open('LCL5_voted.vcf','w') | |||
| benchmark_LCL6 = open('LCL6_voted.vcf','w') | |||
| benchmark_LCL7 = open('LCL7_voted.vcf','w') | |||
| benchmark_LCL8 = open('LCL8_voted.vcf','w') | |||
| all_sample_outfile = open('all_sample_information.txt','w') | |||
| # write VCF | |||
| LCL5_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5_benchmark_calls\n' | |||
| LCL6_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL6_benchmark_calls\n' | |||
| LCL7_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL7_benchmark_calls\n' | |||
| LCL8_col = '#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL8_benchmark_calls\n' | |||
| benchmark_LCL5.write(vcf_header) | |||
| benchmark_LCL5.write(LCL5_col) | |||
| benchmark_LCL6.write(vcf_header) | |||
| benchmark_LCL6.write(LCL6_col) | |||
| benchmark_LCL7.write(vcf_header) | |||
| benchmark_LCL7.write(LCL7_col) | |||
| benchmark_LCL8.write(vcf_header) | |||
| benchmark_LCL8.write(LCL8_col) | |||
| # all info | |||
| all_info_col = 'CHROM\tPOS\tLCL5_pcr_consensus\tLCL5_pcr_free_consensus\tLCL5_mendelian_num\tLCL5_consensus_call\tLCL5_consensus_alt_seq\tLCL5_alt\tLCL5_dp\tLCL5_detected_num\tLCL6_pcr_consensus\tLCL6_pcr_free_consensus\tLCL6_mendelian_num\tLCL6_consensus_call\tLCL6_consensus_alt_seq\tLCL6_alt\tLCL6_dp\tLCL6_detected_num\tLCL7_pcr_consensus\tLCL7_pcr_free_consensus\tLCL7_mendelian_num\t LCL7_consensus_call\tLCL7_consensus_alt_seq\tLCL7_alt\tLCL7_dp\tLCL7_detected_num\tLCL8_pcr_consensus\tLCL8_pcr_free_consensus\tLCL8_mendelian_num\tLCL8_consensus_call\tLCL8_consensus_alt_seq\tLCL8_alt\tLCL8_dp\tLCL8_detected_num\n' | |||
| all_sample_outfile.write(all_info_col) | |||
| # function | |||
| def decide_by_rep(vcf_list,mendelian_list): | |||
| consensus_rep = '' | |||
| gt = [x.split(':')[0] for x in vcf_list] | |||
| # mendelian consistent? | |||
| mendelian_dict = Counter(mendelian_list) | |||
| highest_mendelian = mendelian_dict.most_common(1) | |||
| candidate_mendelian = highest_mendelian[0][0] | |||
| freq_mendelian = highest_mendelian[0][1] | |||
| if (candidate_mendelian == '1:1:1') and (freq_mendelian >= 2): | |||
| con_loc = [i for i in range(len(mendelian_list)) if mendelian_list[i] == '1:1:1'] | |||
| gt_con = itemgetter(*con_loc)(gt) | |||
| gt_num_dict = Counter(gt_con) | |||
| highest_gt = gt_num_dict.most_common(1) | |||
| candidate_gt = highest_gt[0][0] | |||
| freq_gt = highest_gt[0][1] | |||
| if (candidate_gt != './.') and (freq_gt >= 2): | |||
| consensus_rep = candidate_gt | |||
| elif (candidate_gt == './.') and (freq_gt >= 2): | |||
| consensus_rep = 'noGTInfo' | |||
| else: | |||
| consensus_rep = 'inconGT' | |||
| elif (candidate_mendelian == 'nan') and (freq_mendelian >= 2): | |||
| consensus_rep = 'noMenInfo' | |||
| else: | |||
| consensus_rep = 'inconMen' | |||
| return consensus_rep | |||
| def consensus_call(vcf_info_list,mendelian_list,alt_seq): | |||
| pcr_consensus = '.' | |||
| pcr_free_consensus = '.' | |||
| mendelian_num = '.' | |||
| consensus_call = '.' | |||
| consensus_alt_seq = '.' | |||
| # pcr | |||
| SEQ2000 = decide_by_rep(vcf_info_list[0:3],mendelian_list[0:3]) | |||
| XTen_ARD = decide_by_rep(vcf_info_list[18:21],mendelian_list[18:21]) | |||
| XTen_NVG = decide_by_rep(vcf_info_list[21:24],mendelian_list[21:24]) | |||
| XTen_WUX = decide_by_rep(vcf_info_list[24:27],mendelian_list[24:27]) | |||
| pcr_sequence_site = [SEQ2000,XTen_ARD,XTen_NVG,XTen_WUX] | |||
| pcr_sequence_dict = Counter(pcr_sequence_site) | |||
| pcr_highest_sequence = pcr_sequence_dict.most_common(1) | |||
| pcr_candidate_sequence = pcr_highest_sequence[0][0] | |||
| pcr_freq_sequence = pcr_highest_sequence[0][1] | |||
| if pcr_freq_sequence > 2: | |||
| pcr_consensus = pcr_candidate_sequence | |||
| else: | |||
| pcr_consensus = 'inconSequenceSite' | |||
| # pcr-free | |||
| T7_WGE = decide_by_rep(vcf_info_list[3:6],mendelian_list[3:6]) | |||
| Nova_ARD_1 = decide_by_rep(vcf_info_list[6:9],mendelian_list[6:9]) | |||
| Nova_ARD_2 = decide_by_rep(vcf_info_list[9:12],mendelian_list[9:12]) | |||
| Nova_BRG = decide_by_rep(vcf_info_list[12:15],mendelian_list[12:15]) | |||
| Nova_WUX = decide_by_rep(vcf_info_list[15:18],mendelian_list[15:18]) | |||
| sequence_site = [T7_WGE,Nova_ARD_1,Nova_ARD_2,Nova_BRG,Nova_WUX] | |||
| sequence_dict = Counter(sequence_site) | |||
| highest_sequence = sequence_dict.most_common(1) | |||
| candidate_sequence = highest_sequence[0][0] | |||
| freq_sequence = highest_sequence[0][1] | |||
| if freq_sequence > 3: | |||
| pcr_free_consensus = candidate_sequence | |||
| else: | |||
| pcr_free_consensus = 'inconSequenceSite' | |||
| # net alt, dp | |||
| # alt | |||
| AD = [x.split(':')[1] for x in vcf_info_list] | |||
| ALT = [x.split(',')[1] for x in AD] | |||
| ALT = [int(x) for x in ALT] | |||
| ALL_ALT = sum(ALT) | |||
| # dp | |||
| DP = [x.split(':')[2] for x in vcf_info_list] | |||
| DP = [int(x) for x in DP] | |||
| ALL_DP = sum(DP) | |||
| # detected number | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| gt = [x.replace('0/0','.') for x in gt] | |||
| gt = [x.replace('./.','.') for x in gt] | |||
| detected_num = 27 - gt.count('.') | |||
| # decide consensus calls | |||
| tag = ['inconGT','noMenInfo','inconMen','inconSequenceSite','noGTInfo'] | |||
| if (pcr_consensus != '0/0') and (pcr_consensus == pcr_free_consensus) and (pcr_consensus not in tag): | |||
| consensus_call = pcr_consensus | |||
| gt = [x.split(':')[0] for x in vcf_info_list] | |||
| indices = [i for i, x in enumerate(gt) if x == consensus_call] | |||
| matched_mendelian = itemgetter(*indices)(mendelian_list) | |||
| mendelian_num = matched_mendelian.count('1:1:1') | |||
| # Delete multiple alternative genotype to necessary expression | |||
| alt_gt = alt_seq.split(',') | |||
| if len(alt_gt) > 1: | |||
| allele1 = consensus_call.split('/')[0] | |||
| allele2 = consensus_call.split('/')[1] | |||
| if allele1 == '0': | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| consensus_alt_seq = allele2_seq | |||
| consensus_call = '0/1' | |||
| else: | |||
| allele1_seq = alt_gt[int(allele1) - 1] | |||
| allele2_seq = alt_gt[int(allele2) - 1] | |||
| if int(allele1) > int(allele2): | |||
| consensus_alt_seq = allele2_seq + ',' + allele1_seq | |||
| consensus_call = '1/2' | |||
| elif int(allele1) < int(allele2): | |||
| consensus_alt_seq = allele1_seq + ',' + allele2_seq | |||
| consensus_call = '1/2' | |||
| else: | |||
| consensus_alt_seq = allele1_seq | |||
| consensus_call = '1/1' | |||
| else: | |||
| consensus_alt_seq = alt_seq | |||
| elif (pcr_consensus in tag) and (pcr_free_consensus in tag): | |||
| consensus_call = 'filtered' | |||
| elif ((pcr_consensus == './.') or (pcr_consensus in tag)) and ((pcr_free_consensus not in tag) and (pcr_free_consensus != './.')): | |||
| consensus_call = 'pcr-free-speicifc' | |||
| elif ((pcr_consensus != './.') or (pcr_consensus not in tag)) and ((pcr_free_consensus in tag) and (pcr_free_consensus == './.')): | |||
| consensus_call = 'pcr-speicifc' | |||
| elif (pcr_consensus == '0/0') and (pcr_free_consensus == '0/0'): | |||
| consensus_call = '0/0' | |||
| else: | |||
| consensus_call = 'filtered' | |||
| return pcr_consensus, pcr_free_consensus, mendelian_num, consensus_call, consensus_alt_seq, ALL_ALT, ALL_DP, detected_num | |||
| for row in merged_df.itertuples(): | |||
| mendelian_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_1_20191105,row.Quartet_DNA_BGI_T7_WGE_2_20191105,row.Quartet_DNA_BGI_T7_WGE_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_3_20170216] | |||
| lcl5_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL5_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL5_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL5_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL5_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL5_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL5_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL5_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL5_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL5_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL5_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL5_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL5_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL5_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL5_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL5_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL5_3_20170216] | |||
| lcl6_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL6_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL6_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL6_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL6_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL6_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL6_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL6_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL6_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL6_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL6_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL6_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL6_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL6_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL6_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL6_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL6_3_20170216] | |||
| lcl7_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL7_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL7_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL7_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL7_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL7_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL7_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL7_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL7_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL7_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL7_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL7_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL7_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL7_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL7_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL7_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL7_3_20170216] | |||
| lcl8_list = [row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_1_20180518,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_2_20180530,row.Quartet_DNA_BGI_SEQ2000_BGI_LCL8_3_20180530, \ | |||
| row.Quartet_DNA_BGI_T7_WGE_LCL8_1_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_2_20191105,row.Quartet_DNA_BGI_T7_WGE_LCL8_3_20191105, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_1_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_2_20181108,row.Quartet_DNA_ILM_Nova_ARD_LCL8_3_20181108, \ | |||
| row.Quartet_DNA_ILM_Nova_ARD_LCL8_4_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_5_20190111,row.Quartet_DNA_ILM_Nova_ARD_LCL8_6_20190111, \ | |||
| row.Quartet_DNA_ILM_Nova_BRG_LCL8_1_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_2_20180930,row.Quartet_DNA_ILM_Nova_BRG_LCL8_3_20180930, \ | |||
| row.Quartet_DNA_ILM_Nova_WUX_LCL8_1_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_2_20190917,row.Quartet_DNA_ILM_Nova_WUX_LCL8_3_20190917, \ | |||
| row.Quartet_DNA_ILM_XTen_ARD_LCL8_1_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_2_20170403,row.Quartet_DNA_ILM_XTen_ARD_LCL8_3_20170403, \ | |||
| row.Quartet_DNA_ILM_XTen_NVG_LCL8_1_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_2_20170329,row.Quartet_DNA_ILM_XTen_NVG_LCL8_3_20170329, \ | |||
| row.Quartet_DNA_ILM_XTen_WUX_LCL8_1_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_2_20170216,row.Quartet_DNA_ILM_XTen_WUX_LCL8_3_20170216] | |||
| # LCL5 | |||
| LCL5_pcr_consensus, LCL5_pcr_free_consensus, LCL5_mendelian_num, LCL5_consensus_call, LCL5_consensus_alt_seq, LCL5_alt, LCL5_dp, LCL5_detected_num = consensus_call(lcl5_list,mendelian_list,row.ALT) | |||
| if LCL5_mendelian_num != '.': | |||
| LCL5_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL5_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL5_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL5_consensus_call + ':' + str(LCL5_alt) + ':' + str(LCL5_dp) + '\n' | |||
| benchmark_LCL5.write(LCL5_output) | |||
| # LCL6 | |||
| LCL6_pcr_consensus, LCL6_pcr_free_consensus, LCL6_mendelian_num, LCL6_consensus_call, LCL6_consensus_alt_seq, LCL6_alt, LCL6_dp, LCL6_detected_num = consensus_call(lcl6_list,mendelian_list,row.ALT) | |||
| if LCL6_mendelian_num != '.': | |||
| LCL6_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL6_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL6_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL6_consensus_call + ':' + str(LCL6_alt) + ':' + str(LCL6_dp) + '\n' | |||
| benchmark_LCL6.write(LCL6_output) | |||
| # LCL7 | |||
| LCL7_pcr_consensus, LCL7_pcr_free_consensus, LCL7_mendelian_num, LCL7_consensus_call, LCL7_consensus_alt_seq, LCL7_alt, LCL7_dp, LCL7_detected_num = consensus_call(lcl7_list,mendelian_list,row.ALT) | |||
| if LCL7_mendelian_num != '.': | |||
| LCL7_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL7_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL7_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL7_consensus_call + ':' + str(LCL7_alt) + ':' + str(LCL7_dp) + '\n' | |||
| benchmark_LCL7.write(LCL7_output) | |||
| # LCL8 | |||
| LCL8_pcr_consensus, LCL8_pcr_free_consensus, LCL8_mendelian_num, LCL8_consensus_call, LCL8_consensus_alt_seq, LCL8_alt, LCL8_dp, LCL8_detected_num = consensus_call(lcl8_list,mendelian_list,row.ALT) | |||
| if LCL8_mendelian_num != '.': | |||
| LCL8_output = row.CHROM + '\t' + str(row.POS) + '\t' + '.' + '\t' + row.REF + '\t' + LCL8_consensus_alt_seq + '\t' + '.' + '\t' + '.' + '\t' +'VOTED=' + str(LCL8_mendelian_num) + '\t' + 'GT:ALT:DP' + '\t' + LCL8_consensus_call + ':' + str(LCL8_alt) + ':' + str(LCL8_dp) + '\n' | |||
| benchmark_LCL8.write(LCL8_output) | |||
| # all data | |||
| all_output = row.CHROM + '\t' + str(row.POS) + '\t' + LCL5_pcr_consensus + '\t' + LCL5_pcr_free_consensus + '\t' + str(LCL5_mendelian_num) + '\t' + LCL5_consensus_call + '\t' + LCL5_consensus_alt_seq + '\t' + str(LCL5_alt) + '\t' + str(LCL5_dp) + '\t' + str(LCL5_detected_num) + '\t' +\ | |||
| LCL6_pcr_consensus + '\t' + LCL6_pcr_free_consensus + '\t' + str(LCL6_mendelian_num) + '\t' + LCL6_consensus_call + '\t' + LCL6_consensus_alt_seq + '\t' + str(LCL6_alt) + '\t' + str(LCL6_dp) + '\t' + str(LCL6_detected_num) + '\t' +\ | |||
| LCL7_pcr_consensus + '\t' + LCL7_pcr_free_consensus + '\t' + str(LCL7_mendelian_num) + '\t' + LCL7_consensus_call + '\t' + LCL7_consensus_alt_seq + '\t' + str(LCL7_alt) + '\t' + str(LCL7_dp) + '\t' + str(LCL7_detected_num) + '\t' +\ | |||
| LCL8_pcr_consensus + '\t' + LCL8_pcr_free_consensus + '\t' + str(LCL8_mendelian_num) + '\t' + LCL8_consensus_call + '\t' + LCL8_consensus_alt_seq + '\t' + str(LCL8_alt) + '\t' + str(LCL8_dp) + '\t' + str(LCL8_detected_num) + '\n' | |||
| all_sample_outfile.write(all_output) | |||
+ 30
- 0
defaults
Прегледај датотеку
| @@ -0,0 +1,30 @@ | |||
| { | |||
| "benchmarking_dir": "oss://pgx-result/renluyao/manuscript_v3.0/reference_datasets_v202103/", | |||
| "SENTIEON_INSTALL_DIR": "/opt/sentieon-genomics", | |||
| "fasta": "GRCh38.d1.vd1.fa", | |||
| "BENCHMARKdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/rtg-hap:latest", | |||
| "dbsnp_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "BEDTOOLSdocker": "registry-internal.cn-shanghai.aliyuncs.com/pgx-docker-registry/bedtools:v2.27.1", | |||
| "PICARDdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/picard:2.25.4", | |||
| "disk_size": "500", | |||
| "reference_bed_dict": "oss://pgx-reference-data/GRCh38.d1.vd1/GRCh38.d1.vd1.dict", | |||
| "FASTQCdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqc:0.11.8", | |||
| "MULTIQCdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/multiqc:v1.8", | |||
| "SMALLcluster_config": "OnDemand bcs.ps.g.xlarge img-ubuntu-vpc", | |||
| "screen_ref_dir": "oss://pgx-reference-data/fastq_screen_reference/", | |||
| "fastq_1_D5": "", | |||
| "dbmills_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||
| "BIGcluster_config": "OnDemand bcs.a2.7xlarge img-ubuntu-vpc", | |||
| "fastq_screen_conf": "oss://pgx-reference-data/fastq_screen_reference/fastq_screen.conf", | |||
| "FASTQSCREENdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/fastqscreen:0.12.0", | |||
| "SENTIEON_LICENSE": "192.168.0.55:8990", | |||
| "SENTIEONdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/sentieon-genomics:v2019.11.28", | |||
| "QUALIMAPdocker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/qualimap:2.0.0", | |||
| "vcf_D5": "", | |||
| "benchmark_region": "oss://pgx-result/renluyao/manuscript_v3.0/reference_datasets_v202103/Quartet.high.confidence.region.v202103.bed", | |||
| "db_mills": "Mills_and_1000G_gold_standard.indels.hg38.vcf", | |||
| "dbsnp": "dbsnp_146.hg38.vcf", | |||
| "MENDELIANdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/vbt:v1.1", | |||
| "DIYdocker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/high_confidence_call_manuscript:v1.4", | |||
| "ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/" | |||
| } | |||
+ 43
- 0
inputs
Прегледај датотеку
| @@ -0,0 +1,43 @@ | |||
| { | |||
| "{{ project_name }}.benchmarking_dir": "{{ benchmarking_dir }}", | |||
| "{{ project_name }}.vcf_F7": "{{ vcf_F7 }}", | |||
| "{{ project_name }}.SENTIEON_INSTALL_DIR": "{{ SENTIEON_INSTALL_DIR }}", | |||
| "{{ project_name }}.vcf_M8": "{{ vcf_M8 }}", | |||
| "{{ project_name }}.fasta": "{{ fasta }}", | |||
| "{{ project_name }}.BENCHMARKdocker": "{{ BENCHMARKdocker }}", | |||
| "{{ project_name }}.vcf_D6": "{{ vcf_D6 }}", | |||
| "{{ project_name }}.dbsnp_dir": "{{ dbsnp_dir }}", | |||
| "{{ project_name }}.BEDTOOLSdocker": "{{ BEDTOOLSdocker }}", | |||
| "{{ project_name }}.PICARDdocker": "{{ PICARDdocker }}", | |||
| "{{ project_name }}.disk_size": "{{ disk_size }}", | |||
| "{{ project_name }}.FASTQCdocker": "{{ FASTQCdocker }}", | |||
| "{{ project_name }}.MULTIQCdocker": "{{ MULTIQCdocker }}", | |||
| "{{ project_name }}.reference_bed_dict": "{{ reference_bed_dict }}", | |||
| "{{ project_name }}.fastq_2_M8": "{{ fastq_2_M8 }}", | |||
| "{{ project_name }}.project": "{{ project_name }}", | |||
| "{{ project_name }}.pl": "{{ pl }}", | |||
| "{{ project_name }}.bed": "{{ bed }}", | |||
| "{{ project_name }}.fastq_1_M8": "{{ fastq_1_M8 }}", | |||
| "{{ project_name }}.SMALLcluster_config": "{{ SMALLcluster_config }}", | |||
| "{{ project_name }}.screen_ref_dir": "{{ screen_ref_dir }}", | |||
| "{{ project_name }}.fastq_1_D5": "{{ fastq_1_D5 }}", | |||
| "{{ project_name }}.dbmills_dir": "{{ dbmills_dir }}", | |||
| "{{ project_name }}.BIGcluster_config": "{{ BIGcluster_config }}", | |||
| "{{ project_name }}.fastq_screen_conf": "{{ fastq_screen_conf }}", | |||
| "{{ project_name }}.fastq_2_D5": "{{ fastq_2_D5 }}", | |||
| "{{ project_name }}.FASTQSCREENdocker": "{{ FASTQSCREENdocker }}", | |||
| "{{ project_name }}.fastq_2_F7": "{{ fastq_2_F7 }}", | |||
| "{{ project_name }}.SENTIEON_LICENSE": "{{ SENTIEON_LICENSE }}", | |||
| "{{ project_name }}.fastq_1_D6": "{{ fastq_1_D6 }}", | |||
| "{{ project_name }}.fastq_1_F7": "{{ fastq_1_F7 }}", | |||
| "{{ project_name }}.SENTIEONdocker": "{{ SENTIEONdocker }}", | |||
| "{{ project_name }}.QUALIMAPdocker": "{{ QUALIMAPdocker }}", | |||
| "{{ project_name }}.vcf_D5": "{{ vcf_D5 }}", | |||
| "{{ project_name }}.benchmark_region": "{{ benchmark_region }}", | |||
| "{{ project_name }}.db_mills": "{{ db_mills }}", | |||
| "{{ project_name }}.dbsnp": "{{ dbsnp }}", | |||
| "{{ project_name }}.MENDELIANdocker": "{{ MENDELIANdocker }}", | |||
| "{{ project_name }}.fastq_2_D6": "{{ fastq_2_D6 }}", | |||
| "{{ project_name }}.DIYdocker": "{{ DIYdocker }}", | |||
| "{{ project_name }}.ref_dir": "{{ ref_dir }}" | |||
| } | |||
BIN
pictures/Picture1.png
Прегледај датотеку
{kind=link}
| Before | After |
|---|---|
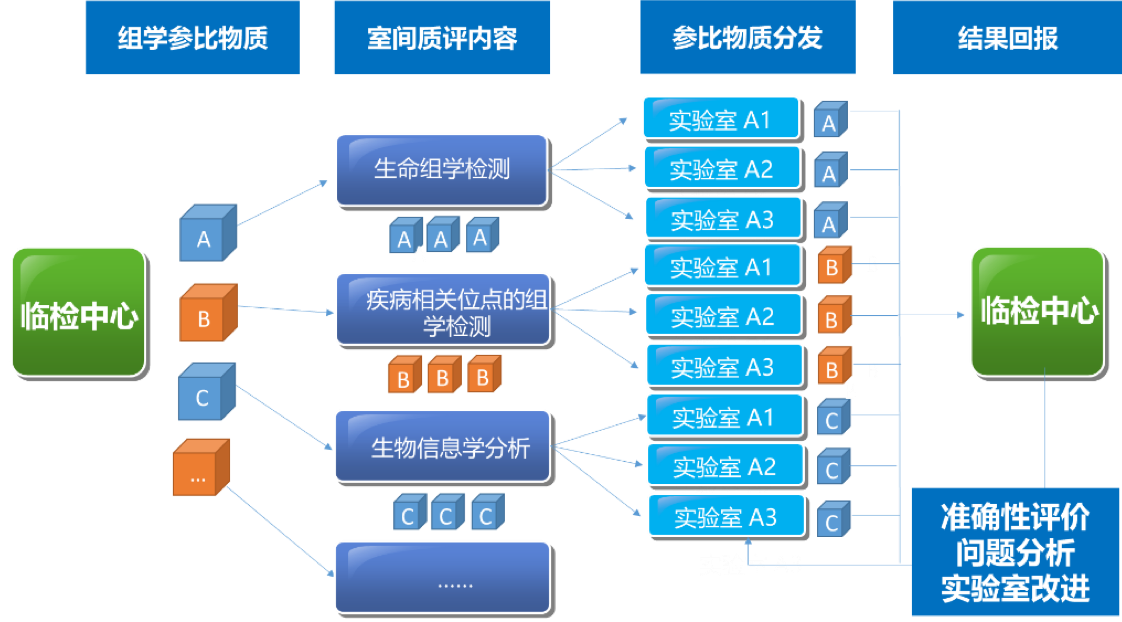
|
|
| Width: 1122 | Height: 622 | Size: 240KB |
BIN
pictures/performance.png
Прегледај датотеку
{kind=link}
| Before | After |
|---|---|
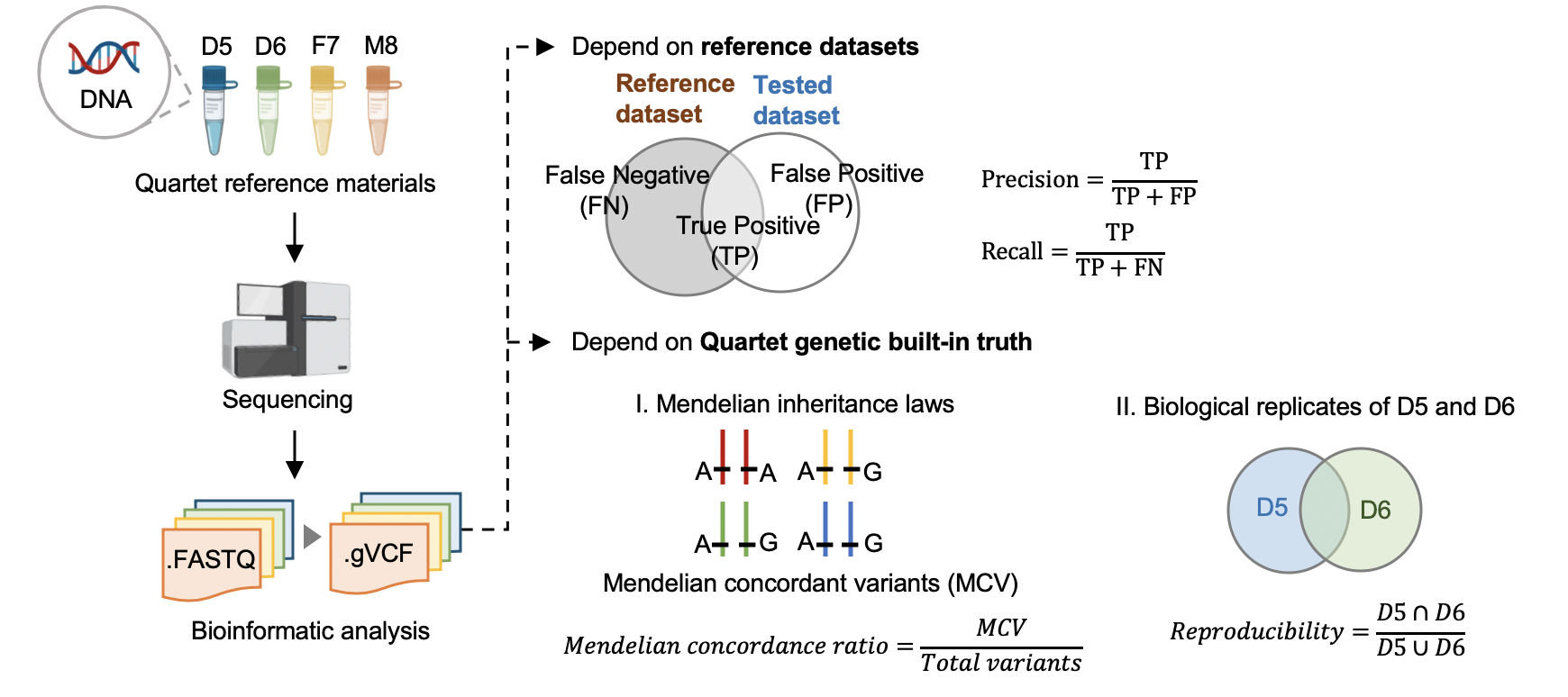
|
|
| Width: 1740 | Height: 774 | Size: 387KB |
BIN
pictures/table.png
Прегледај датотеку
{kind=link}
| Before | After |
|---|---|
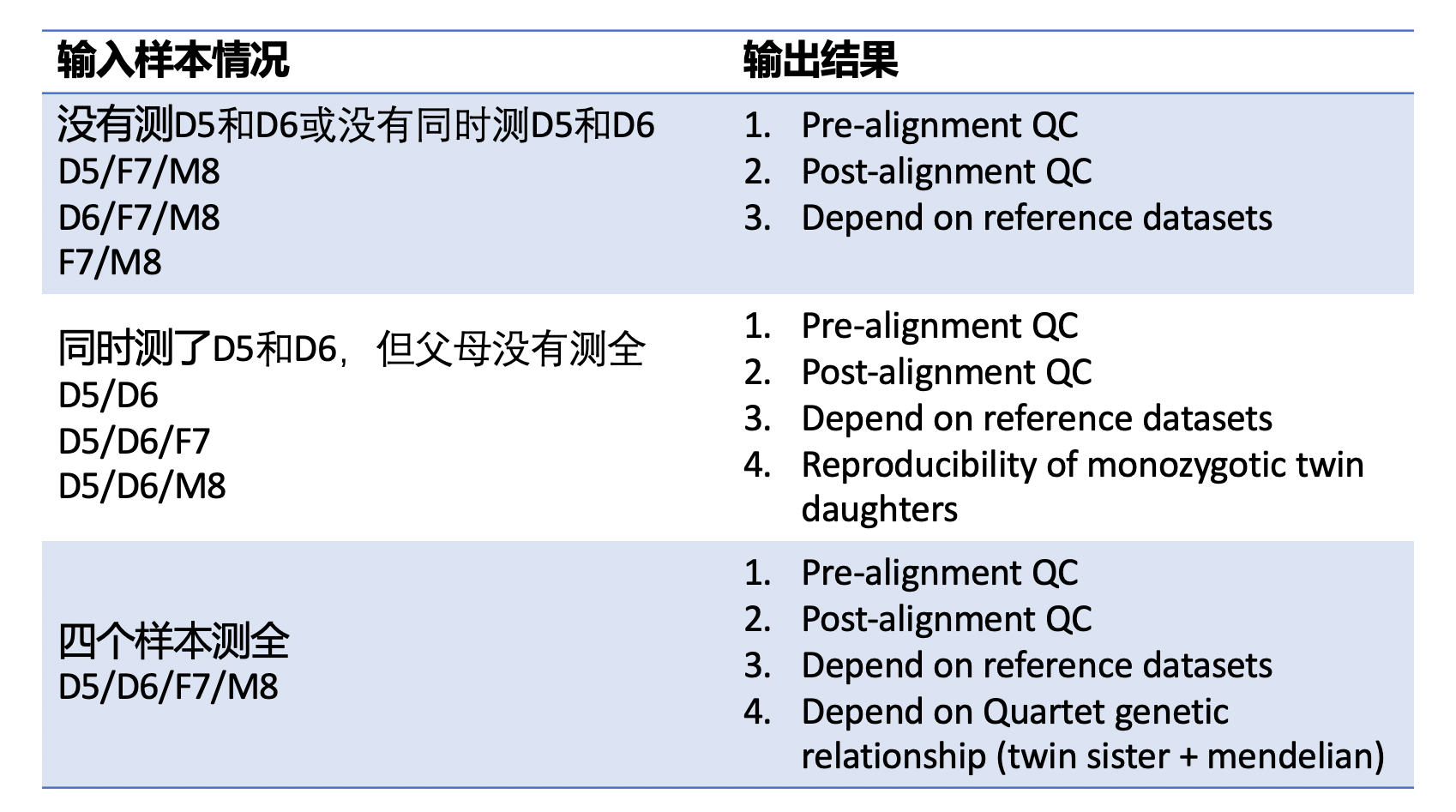
|
|
| Width: 1698 | Height: 947 | Size: 448KB |
BIN
pictures/workflow.png
Прегледај датотеку
{kind=link}
| Before | After |
|---|---|
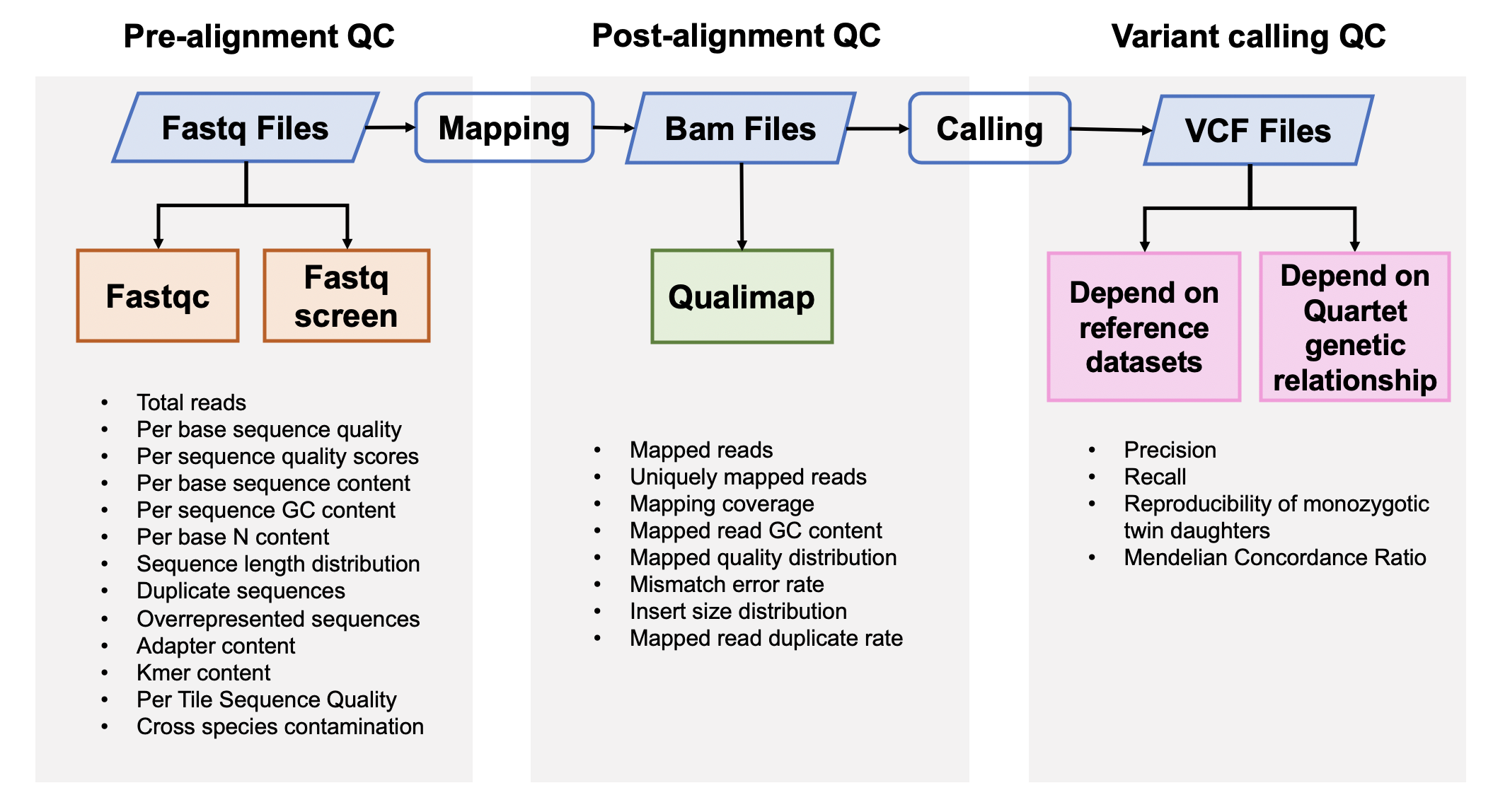
|
|
| Width: 2110 | Height: 1148 | Size: 599KB |
BIN
tasks/.DS_Store
Прегледај датотеку
+ 48
- 0
tasks/BQSR.wdl
Прегледај датотеку
| @@ -0,0 +1,48 @@ | |||
| task BQSR { | |||
| File ref_dir | |||
| File dbsnp_dir | |||
| File dbmills_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| String dbsnp | |||
| String db_mills | |||
| File realigned_bam | |||
| File realigned_bam_index | |||
| String sample = basename(realigned_bam,".sorted.deduped.realigned.bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${realigned_bam} -q ${sample}_recal_data.table --algo QualCal -k ${dbsnp_dir}/${dbsnp} -k ${dbmills_dir}/${db_mills} ${sample}_recal_data.table.post --algo ReadWriter ${sample}.sorted.deduped.realigned.recaled.bam | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt --algo QualCal --plot --before ${sample}_recal_data.table --after ${sample}_recal_data.table.post ${sample}_recal_data.csv | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon plot QualCal -o ${sample}_bqsrreport.pdf ${sample}_recal_data.csv | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File recal_table = "${sample}_recal_data.table" | |||
| File recal_post = "${sample}_recal_data.table.post" | |||
| File recaled_bam = "${sample}.sorted.deduped.realigned.recaled.bam" | |||
| File recaled_bam_index = "${sample}.sorted.deduped.realigned.recaled.bam.bai" | |||
| File recal_csv = "${sample}_recal_data.csv" | |||
| File bqsrreport_pdf = "${sample}_bqsrreport.pdf" | |||
| } | |||
| } | |||
+ 39
- 0
tasks/Dedup.wdl
Прегледај датотеку
| @@ -0,0 +1,39 @@ | |||
| task Dedup { | |||
| String SENTIEON_INSTALL_DIR | |||
| File sorted_bam | |||
| File sorted_bam_index | |||
| String sample = basename(sorted_bam,".sorted.bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo LocusCollector --fun score_info ${sample}_score.txt | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -t $nt -i ${sorted_bam} --algo Dedup --rmdup --score_info ${sample}_score.txt --metrics ${sample}_dedup_metrics.txt ${sample}.sorted.deduped.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File score = "${sample}_score.txt" | |||
| File dedup_metrics = "${sample}_dedup_metrics.txt" | |||
| File Dedup_bam = "${sample}.sorted.deduped.bam" | |||
| File Dedup_bam_index = "${sample}.sorted.deduped.bam.bai" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/Haplotyper.wdl
Прегледај датотеку
| @@ -0,0 +1,34 @@ | |||
| task Haplotyper { | |||
| File ref_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File recaled_bam | |||
| File recaled_bam_index | |||
| String sample = basename(recaled_bam,".sorted.deduped.realigned.recaled.bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper ${sample}_hc.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf = "${sample}_hc.vcf" | |||
| File vcf_idx = "${sample}_hc.vcf.idx" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/Realigner.wdl
Прегледај датотеку
| @@ -0,0 +1,41 @@ | |||
| task Realigner { | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| String sample = basename(Dedup_bam,".sorted.deduped.bam") | |||
| String db_mills | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo Realigner -k ${dbmills_dir}/${db_mills} ${sample}.sorted.deduped.realigned.bam | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File realigner_bam = "${sample}.sorted.deduped.realigned.bam" | |||
| File realigner_bam_index = "${sample}.sorted.deduped.realigned.bam.bai" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/bed_to_interval_list.wdl
Прегледај датотеку
| @@ -0,0 +1,34 @@ | |||
| task bed_to_interval_list { | |||
| File reference_bed_dict | |||
| File bed | |||
| File ref_dir | |||
| String fasta | |||
| String interval_list_name | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| mkdir -p /cromwell_root/tmp | |||
| cp ${ref_dir}/${fasta} /cromwell_root/tmp/ | |||
| cp ${reference_bed_dict} /cromwell_root/tmp/ | |||
| java -jar /usr/picard/picard.jar BedToIntervalList \ | |||
| I=${bed} \ | |||
| O=${interval_list_name}.txt \ | |||
| SD=/cromwell_root/tmp/GRCh38.d1.vd1.dict | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File interval_list = "${interval_list_name}.txt" | |||
| } | |||
| } | |||
+ 84
- 0
tasks/benchmark.wdl
Прегледај датотеку
| @@ -0,0 +1,84 @@ | |||
| task benchmark { | |||
| File filtered_vcf | |||
| File benchmarking_dir | |||
| File ref_dir | |||
| File bed | |||
| String sample = basename(filtered_vcf,".filtered.vcf") | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${ref_dir} /cromwell_root/tmp/ | |||
| cp -r ${benchmarking_dir} /cromwell_root/tmp/ | |||
| export HGREF=/cromwell_root/tmp/reference_data/GRCh38.d1.vd1.fa | |||
| echo -e "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL5" > LCL5_name | |||
| echo -e "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL6" > LCL6_name | |||
| echo -e "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL7" > LCL7_name | |||
| echo -e "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO\tFORMAT\tLCL8" > LCL8_name | |||
| if [[ ${sample} =~ "LCL5" ]];then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL5.high.confidence.calls.vcf ${filtered_vcf} -f ${bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| cat ${filtered_vcf} | grep '##' > header | |||
| cat ${filtered_vcf} | grep -v '#' > body | |||
| cat header LCL5_name body > LCL5.vcf | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip LCL5.vcf -c > ${sample}.reformed.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.reformed.vcf.gz | |||
| elif [[ ${sample} =~ "LCL6" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL6.high.confidence.calls.vcf ${filtered_vcf} -f ${bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| cat ${filtered_vcf} | grep '##' > header | |||
| cat ${filtered_vcf} | grep -v '#' > body | |||
| cat header LCL6_name body > LCL6.vcf | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip LCL6.vcf -c > ${sample}.reformed.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.reformed.vcf.gz | |||
| elif [[ ${sample} =~ "LCL7" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL7.high.confidence.calls.vcf ${filtered_vcf} -f ${bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| cat ${filtered_vcf} | grep '##' > header | |||
| cat ${filtered_vcf} | grep -v '#' > body | |||
| cat header LCL7_name body > LCL7.vcf | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip LCL7.vcf -c > ${sample}.reformed.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.reformed.vcf.gz | |||
| elif [[ ${sample} =~ "LCL8" ]]; then | |||
| /opt/hap.py/bin/hap.py /cromwell_root/tmp/reference_datasets_v202103/LCL8.high.confidence.calls.vcf ${filtered_vcf} -f ${bed} --threads $nt -o ${sample} -r ${ref_dir}/${fasta} | |||
| cat ${filtered_vcf} | grep '##' > header | |||
| cat ${filtered_vcf} | grep -v '#' > body | |||
| cat header LCL8_name body > LCL8.vcf | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg bgzip LCL8.vcf -c > ${sample}.reformed.vcf.gz | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg index -f vcf ${sample}.reformed.vcf.gz | |||
| else | |||
| echo "only for quartet samples" | |||
| fi | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File rtg_vcf = "${sample}.reformed.vcf.gz" | |||
| File rtg_vcf_index = "${sample}.reformed.vcf.gz.tbi" | |||
| File gzip_vcf = "${sample}.vcf.gz" | |||
| File gzip_vcf_index = "${sample}.vcf.gz.tbi" | |||
| File roc_all_csv = "${sample}.roc.all.csv.gz" | |||
| File roc_indel = "${sample}.roc.Locations.INDEL.csv.gz" | |||
| File roc_indel_pass = "${sample}.roc.Locations.INDEL.PASS.csv.gz" | |||
| File roc_snp = "${sample}.roc.Locations.SNP.csv.gz" | |||
| File roc_snp_pass = "${sample}.roc.Locations.SNP.PASS.csv.gz" | |||
| File summary = "${sample}.summary.csv" | |||
| File extended = "${sample}.extended.csv" | |||
| File metrics = "${sample}.metrics.json.gz" | |||
| } | |||
| } | |||
+ 47
- 0
tasks/deduped_Metrics.wdl
Прегледај датотеку
| @@ -0,0 +1,47 @@ | |||
| task deduped_Metrics { | |||
| File ref_dir | |||
| File bed | |||
| String SENTIEON_INSTALL_DIR | |||
| String fasta | |||
| File Dedup_bam | |||
| File Dedup_bam_index | |||
| File interval_list | |||
| String sample = basename(Dedup_bam,".sorted.deduped.bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=192.168.0.55:8990 | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --interval ${bed} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt --algo QualityYield ${sample}_deduped_QualityYield.txt --algo WgsMetricsAlgo ${sample}_deduped_WgsMetricsAlgo.txt --algo HsMetricAlgo --targets_list ${interval_list} --baits_list ${interval_list} ${sample}_deduped_HsMetricAlgo.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File deduped_coverage_metrics_sample_summary = "${sample}_deduped_coverage_metrics.sample_summary" | |||
| File deduped_coverage_metrics_sample_statistics = "${sample}_deduped_coverage_metrics.sample_statistics" | |||
| File deduped_coverage_metrics_sample_interval_statistics = "${sample}_deduped_coverage_metrics.sample_interval_statistics" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_proportions = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_proportions" | |||
| File deduped_coverage_metrics_sample_cumulative_coverage_counts = "${sample}_deduped_coverage_metrics.sample_cumulative_coverage_counts" | |||
| File deduped_mean_quality = "${sample}_deduped_mq_metrics.txt" | |||
| File deduped_qd_metrics = "${sample}_deduped_qd_metrics.txt" | |||
| File deduped_gc_summary = "${sample}_deduped_gc_summary.txt" | |||
| File deduped_gc_metrics = "${sample}_deduped_gc_metrics.txt" | |||
| File dedeuped_aln_metrics = "${sample}_deduped_aln_metrics.txt" | |||
| File deduped_is_metrics = "${sample}_deduped_is_metrics.txt" | |||
| File deduped_QualityYield = "${sample}_deduped_QualityYield.txt" | |||
| File deduped_wgsmetrics = "${sample}_deduped_WgsMetricsAlgo.txt" | |||
| File deduped_hsmetrics = "${sample}_deduped_HsMetricAlgo.txt" | |||
| } | |||
| } | |||
+ 34
- 0
tasks/extract_tables.wdl
Прегледај датотеку
| @@ -0,0 +1,34 @@ | |||
| task extract_tables { | |||
| File quality_yield_summary | |||
| File wgs_metrics_summary | |||
| File aln_metrics_summary | |||
| File is_metrics_summary | |||
| File hs_metrics_summary | |||
| File hap | |||
| File fastqc | |||
| File fastqscreen | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/extract_tables.py -quality ${quality_yield_summary} -depth ${wgs_metrics_summary} -aln ${aln_metrics_summary} -is ${is_metrics_summary} -fastqc ${fastqc} -fastqscreen ${fastqscreen} -hap ${hap} -project ${project} -hs {hs_metrics_summary} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File pre_alignment = "pre_alignment.txt" | |||
| File post_alignment = "post_alignment.txt" | |||
| File variant_calling = "variants.calling.qc.txt" | |||
| } | |||
| } | |||
+ 23
- 0
tasks/extract_tables_vcf.wdl
Прегледај датотеку
| @@ -0,0 +1,23 @@ | |||
| task extract_tables_vcf { | |||
| File hap | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| python /opt/extract_tables.py -hap ${hap} -project ${project} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File variant_calling = "variants.calling.qc.txt" | |||
| } | |||
| } | |||
+ 33
- 0
tasks/fastqc.wdl
Прегледај датотеку
| @@ -0,0 +1,33 @@ | |||
| task fastqc { | |||
| File read1 | |||
| File read2 | |||
| String docker | |||
| String project | |||
| String sample | |||
| String user_define_name = sub(basename(read1, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| cp ${read1} ${user_define_name}_${project}_${sample}_R1.fastq.gz | |||
| cp ${read2} ${user_define_name}_${project}_${sample}_R2.fastq.gz | |||
| fastqc -t $nt -o ./ ${user_define_name}_${project}_${sample}_R1.fastq.gz | |||
| fastqc -t $nt -o ./ ${user_define_name}_${project}_${sample}_R2.fastq.gz | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File read1_html = "${user_define_name}_${project}_${sample}_R1_fastqc.html" | |||
| File read1_zip = "${user_define_name}_${project}_${sample}_R1_fastqc.zip" | |||
| File read2_html = "${user_define_name}_${project}_${sample}_R2_fastqc.html" | |||
| File read2_zip = "${user_define_name}_${project}_${sample}_R2_fastqc.zip" | |||
| } | |||
| } | |||
+ 40
- 0
tasks/fastqscreen.wdl
Прегледај датотеку
| @@ -0,0 +1,40 @@ | |||
| task fastq_screen { | |||
| File read1 | |||
| File read2 | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| String docker | |||
| String project | |||
| String user_define_name = sub(basename(read1, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| String sample | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| mkdir -p /cromwell_root/tmp | |||
| cp -r ${screen_ref_dir} /cromwell_root/tmp/ | |||
| cp ${read1} ${user_define_name}_${project}_${sample}_R1.fastq.gz | |||
| cp ${read2} ${user_define_name}_${project}_${sample}_R2.fastq.gz | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --subset 1000000 --threads $nt ${user_define_name}_${project}_${sample}_R1.fastq.gz | |||
| fastq_screen --aligner bowtie2 --conf ${fastq_screen_conf} --subset 1000000 --threads $nt ${user_define_name}_${project}_${sample}_R2.fastq.gz | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File png1 = "${user_define_name}_${project}_${sample}_R1_screen.png" | |||
| File txt1 = "${user_define_name}_${project}_${sample}_R1_screen.txt" | |||
| File html1 = "${user_define_name}_${project}_${sample}_R1_screen.html" | |||
| File png2 = "${user_define_name}_${project}_${sample}_R2_screen.png" | |||
| File txt2 = "${user_define_name}_${project}_${sample}_R2_screen.txt" | |||
| File html2 = "${user_define_name}_${project}_${sample}_R2_screen.html" | |||
| } | |||
| } | |||
+ 33
- 0
tasks/filter_vcf.wdl
Прегледај датотеку
| @@ -0,0 +1,33 @@ | |||
| task filter_vcf { | |||
| File vcf | |||
| File bed | |||
| File benchmark_region | |||
| String project | |||
| String sample = basename(vcf,".vcf") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${vcf} | grep '#' > header | |||
| cat ${vcf} | grep -v '#' > body | |||
| cat body | grep -w '^chr1\|^chr2\|^chr3\|^chr4\|^chr5\|^chr6\|^chr7\|^chr8\|^chr9\|^chr10\|^chr11\|^chr12\|^chr13\|^chr14\|^chr15\|^chr16\|^chr17\|^chr18\|^chr19\|^chr20\|^chr21\|^chr22\|^chrX' > body.filtered | |||
| cat header body.filtered > ${sample}.filtered.vcf | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${sample}.filtered.vcf -b ${bed} > body.bed.filtered | |||
| cat header body.bed.filtered > ${project}.${sample}.chrom.bed.filtered.vcf | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${benchmark_region} -b ${bed} > benchmark_region_query_bed.bed | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File filtered_vcf = "${project}.${sample}.chrom.bed.filtered.vcf" | |||
| File filtered_bed = "benchmark_region_query_bed.bed" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/filter_vcf_bed.wdl
Прегледај датотеку
| @@ -0,0 +1,37 @@ | |||
| task filter_vcf_bed { | |||
| File vcf | |||
| File? bed | |||
| File benchmark_region | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${vcf} | grep '#' > header | |||
| cat ${vcf} | grep -v '#' > body | |||
| cat body | grep -w '^chr1\|^chr2\|^chr3\|^chr4\|^chr5\|^chr6\|^chr7\|^chr8\|^chr9\|^chr10\|^chr11\|^chr12\|^chr13\|^chr14\|^chr15\|^chr16\|^chr17\|^chr18\|^chr19\|^chr20\|^chr21\|^chr22\|^chrX' > body.filtered | |||
| cat header body.filtered > ${project}.filtered.vcf | |||
| if [ ${bed} ];then | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${project}.filtered.vcf -b ${bed} > body.bed.filtered | |||
| cat header body.bed.filtered > ${project}.filtered.vcf | |||
| /opt/ccdg/bedtools-2.27.1/bin/bedtools intersect -a ${benchmark_region} -b ${bed} > benchmark_region_query_bed.bed | |||
| fi | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File filtered_vcf = "${project}.filtered.vcf" | |||
| Array[File] filtered_bed = glob("benchmark_region_query_bed.bed") | |||
| } | |||
| } | |||
+ 46
- 0
tasks/get_variants_in_del.wdl
Прегледај датотеку
| @@ -0,0 +1,46 @@ | |||
| task adjust_mendelian_status_del { | |||
| File D5_trio_vcf | |||
| File D6_trio_vcf | |||
| File del_bed | |||
| String family_name = basename(D5_trio_vcf,".D5.vcf" | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||
| nt=$(nproc) | |||
| echo -e "${family_name}\tM8\t0\t0\t2\t-9\n${family_name}\tF7\t0\t0\t1\t-9\n${family_name}\tD5\tF7\tM8\t2\t-9" > ${family_name}.D5.ped | |||
| mkdir VBT_D5 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D5.ped -outDir VBT_D5 -out-prefix ${family_name}.D5 --output-violation-regions -thread-count $nt | |||
| cat VBT_D5/${family_name}.D5_trio.vcf > ${family_name}.D5.vcf | |||
| echo -e "${family_name}\tM8\t0\t0\t2\t-9\n${family_name}\tF7\t0\t0\t1\t-9\n${family_name}\tD6\tF7\tM8\t2\t-9" > ${family_name}.D6.ped | |||
| mkdir VBT_D6 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D6.ped -outDir VBT_D6 -out-prefix ${family_name}.D6 --output-violation-regions -thread-count $nt | |||
| cat VBT_D6/${family_name}.D6_trio.vcf > ${family_name}.D6.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File D5_ped = "${family_name}.D5.ped" | |||
| File D6_ped = "${family_name}.D6.ped" | |||
| Array[File] D5_mendelian = glob("VBT_D5/*") | |||
| Array[File] D6_mendelian = glob("VBT_D6/*") | |||
| File D5_trio_vcf = "${family_name}.D5.vcf" | |||
| File D6_trio_vcf = "${family_name}.D6.vcf" | |||
| } | |||
| } | |||
+ 37
- 0
tasks/mapping.wdl
Прегледај датотеку
| @@ -0,0 +1,37 @@ | |||
| task mapping { | |||
| File ref_dir | |||
| String fasta | |||
| File fastq_1 | |||
| File fastq_2 | |||
| String SENTIEON_INSTALL_DIR | |||
| String SENTIEON_LICENSE | |||
| String group | |||
| String sample | |||
| String project | |||
| String pl | |||
| String user_define_name = sub(basename(fastq_1, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| export SENTIEON_LICENSE=${SENTIEON_LICENSE} | |||
| nt=$(nproc) | |||
| ${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${user_define_name}_${project}_${sample}.sorted.bam -t $nt --sam2bam -i - | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File sorted_bam = "${user_define_name}_${project}_${sample}.sorted.bam" | |||
| File sorted_bam_index = "${user_define_name}_${project}_${sample}.sorted.bam.bai" | |||
| } | |||
| } | |||
+ 46
- 0
tasks/mendelian.wdl
Прегледај датотеку
| @@ -0,0 +1,46 @@ | |||
| task mendelian { | |||
| File family_vcf | |||
| File ref_dir | |||
| String family_name = basename(family_vcf,".family.vcf") | |||
| String fasta | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| export LD_LIBRARY_PATH=/opt/htslib-1.9 | |||
| nt=$(nproc) | |||
| echo -e "${family_name}\tLCL8\t0\t0\t2\t-9\n${family_name}\tLCL7\t0\t0\t1\t-9\n${family_name}\tLCL5\tLCL7\tLCL8\t2\t-9" > ${family_name}.D5.ped | |||
| mkdir VBT_D5 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D5.ped -outDir VBT_D5 -out-prefix ${family_name}.D5 --output-violation-regions -thread-count $nt | |||
| cat VBT_D5/${family_name}.D5_trio.vcf > ${family_name}.D5.vcf | |||
| echo -e "${family_name}\tLCL8\t0\t0\t2\t-9\n${family_name}\tLCL7\t0\t0\t1\t-9\n${family_name}\tLCL6\tLCL7\tLCL8\t2\t-9" > ${family_name}.D6.ped | |||
| mkdir VBT_D6 | |||
| /opt/VBT-TrioAnalysis/vbt mendelian -ref ${ref_dir}/${fasta} -mother ${family_vcf} -father ${family_vcf} -child ${family_vcf} -pedigree ${family_name}.D6.ped -outDir VBT_D6 -out-prefix ${family_name}.D6 --output-violation-regions -thread-count $nt | |||
| cat VBT_D6/${family_name}.D6_trio.vcf > ${family_name}.D6.vcf | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File D5_ped = "${family_name}.D5.ped" | |||
| File D6_ped = "${family_name}.D6.ped" | |||
| Array[File] D5_mendelian = glob("VBT_D5/*") | |||
| Array[File] D6_mendelian = glob("VBT_D6/*") | |||
| File D5_trio_vcf = "${family_name}.D5.vcf" | |||
| File D6_trio_vcf = "${family_name}.D6.vcf" | |||
| } | |||
| } | |||
+ 31
- 0
tasks/merge_family.wdl
Прегледај датотеку
| @@ -0,0 +1,31 @@ | |||
| task merge_family { | |||
| File D5_vcf | |||
| File D6_vcf | |||
| File F7_vcf | |||
| File M8_vcf | |||
| File D5_vcf_tbi | |||
| File D6_vcf_tbi | |||
| File F7_vcf_tbi | |||
| File M8_vcf_tbi | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| /opt/rtg-tools/dist/rtg-tools-3.10.1-4d58ead/rtg vcfmerge --force-merge-all -o ${project}.family.vcf.gz ${D5_vcf} ${D6_vcf} ${F7_vcf} ${M8_vcf} | |||
| gunzip ${project}.family.vcf.gz | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File family_vcf = "${project}.family.vcf" | |||
| } | |||
| } | |||
+ 35
- 0
tasks/merge_mendelian.wdl
Прегледај датотеку
| @@ -0,0 +1,35 @@ | |||
| task merge_mendelian { | |||
| File D5_trio_vcf | |||
| File D6_trio_vcf | |||
| File family_vcf | |||
| String family_name = basename(family_vcf,".family.vcf") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cat ${D5_trio_vcf} | grep -v '##' > ${family_name}.D5.txt | |||
| cat ${D6_trio_vcf} | grep -v '##' > ${family_name}.D6.txt | |||
| cat ${family_vcf} | grep -v '##' | awk ' | |||
| BEGIN { OFS = "\t" } | |||
| NF > 2 && FNR > 1 { | |||
| for ( i=9; i<=NF; i++ ) { | |||
| split($i,a,":") ;$i = a[1]; | |||
| } | |||
| } | |||
| { print } | |||
| ' > ${family_name}.consensus.txt | |||
| python /opt/merge_two_family_with_genotype.py -LCL5 ${family_name}.D5.txt -LCL6 ${family_name}.D6.txt -genotype ${family_name}.consensus.txt -family ${family_name} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File project_mendelian = "${family_name}.txt" | |||
| File project_mendelian_summary = "${family_name}.summary.txt" | |||
| } | |||
| } | |||
+ 52
- 0
tasks/merge_sentieon_metrics.wdl
Прегледај датотеку
| @@ -0,0 +1,52 @@ | |||
| task merge_sentieon_metrics { | |||
| Array[File] quality_yield_header | |||
| Array[File] wgs_metrics_algo_header | |||
| Array[File] aln_metrics_header | |||
| Array[File] is_metrics_header | |||
| Array[File] hs_metrics_header | |||
| Array[File] quality_yield_data | |||
| Array[File] wgs_metrics_algo_data | |||
| Array[File] aln_metrics_data | |||
| Array[File] is_metrics_data | |||
| Array[File] hs_metrics_data | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| echo '''Sample''' > sample_column | |||
| cat ${sep=" " quality_yield_header} | sed -n '1,1p' | cat - ${sep=" " quality_yield_data} > quality_yield_all | |||
| ls ${sep=" " quality_yield_data} | cut -d '.' -f1 | cat sample_column - | paste - quality_yield_all > ${project}.quality_yield.txt | |||
| cat ${sep=" " wgs_metrics_algo_header} | sed -n '1,1p' | cat - ${sep=" " wgs_metrics_algo_data} > wgs_metrics_all | |||
| ls ${sep=" " wgs_metrics_algo_data} | cut -d '.' -f1 | cat sample_column - | paste - wgs_metrics_all > ${project}.wgs_metrics_data.txt | |||
| cat ${sep=" " aln_metrics_header} | sed -n '1,1p' | cat - ${sep=" " aln_metrics_data} > aln_metrics_all | |||
| ls ${sep=" " aln_metrics_data} | cut -d '.' -f1 | cat sample_column - | paste - aln_metrics_all > ${project}.aln_metrics.txt | |||
| cat ${sep=" " is_metrics_header} | sed -n '1,1p' | cat - ${sep=" " is_metrics_data} > is_metrics_all | |||
| ls ${sep=" " is_metrics_data} | cut -d '.' -f1 | cat sample_column - | paste - is_metrics_all > ${project}.is_metrics.txt | |||
| cat ${sep=" " hs_metrics_header} | sed -n '1,1p' | cat - ${sep=" " hs_metrics_data} > hs_metrics_all | |||
| ls ${sep=" " hs_metrics_data} | cut -d '.' -f1 | cat sample_column - | paste - hs_metrics_all > ${project}.hs_metrics.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster: cluster_config | |||
| systemDisk: "cloud_ssd 40" | |||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File quality_yield_summary = "${project}.quality_yield.txt" | |||
| File wgs_metrics_summary = "${project}.wgs_metrics_data.txt" | |||
| File aln_metrics_summary = "${project}.aln_metrics.txt" | |||
| File is_metrics_summary = "${project}.is_metrics.txt" | |||
| File hs_metrics_summary = "${project.hs_metrics.txt" | |||
| } | |||
| } | |||
+ 48
- 0
tasks/multiqc.wdl
Прегледај датотеку
| @@ -0,0 +1,48 @@ | |||
| task multiqc { | |||
| Array[File] read1_zip | |||
| Array[File] read2_zip | |||
| Array[File] txt1 | |||
| Array[File] txt2 | |||
| Array[File] summary | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| mkdir -p /cromwell_root/tmp/fastqc | |||
| mkdir -p /cromwell_root/tmp/fastqscreen | |||
| mkdir -p /cromwell_root/tmp/benchmark | |||
| cp ${sep=" " read1_zip} ${sep=" " read2_zip} /cromwell_root/tmp/fastqc | |||
| cp ${sep=" " txt1} ${sep=" " txt2} /cromwell_root/tmp/fastqscreen | |||
| cp ${sep=" " summary} /cromwell_root/tmp/benchmark | |||
| multiqc /cromwell_root/tmp/ | |||
| cat multiqc_data/multiqc_general_stats.txt > multiqc_general_stats.txt | |||
| cat multiqc_data/multiqc_fastq_screen.txt > multiqc_fastq_screen.txt | |||
| cat multiqc_data/multiqc_happy_data.json > multiqc_happy_data.json | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File multiqc_html = "multiqc_report.html" | |||
| Array[File] multiqc_txt = glob("multiqc_data/*") | |||
| File? fastqc = "multiqc_general_stats.txt" | |||
| File? fastqscreen = "multiqc_fastq_screen.txt" | |||
| File hap = "multiqc_happy_data.json" | |||
| } | |||
| } | |||
+ 31
- 0
tasks/multiqc_hap.wdl
Прегледај датотеку
| @@ -0,0 +1,31 @@ | |||
| task multiqc_hap { | |||
| Array[File] summary | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| mkdir -p /cromwell_root/tmp/benchmark | |||
| cp ${sep=" " summary} /cromwell_root/tmp/benchmark | |||
| multiqc /cromwell_root/tmp/ | |||
| cat multiqc_data/multiqc_happy_data.json > multiqc_happy_data.json | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File multiqc_html = "multiqc_report.html" | |||
| Array[File] multiqc_txt = glob("multiqc_data/*") | |||
| File hap = "multiqc_happy_data.json" | |||
| } | |||
| } | |||
+ 30
- 0
tasks/qualimap.wdl
Прегледај датотеку
| @@ -0,0 +1,30 @@ | |||
| task qualimap { | |||
| File bam | |||
| File bai | |||
| File bed | |||
| String bamname = basename(bam,".bam") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| nt=$(nproc) | |||
| awk 'BEGIN{OFS="\t"}{sub("\r","",$3);print $1,$2,$3,"",0,"."}' ${bed} > new.bed | |||
| /opt/qualimap/qualimap bamqc -bam ${bam} -gff new.bed -outformat PDF:HTML -nt $nt -outdir ${bamname} --java-mem-size=60G | |||
| tar -zcvf ${bamname}_qualimap.zip ${bamname} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File zip = "${bamname}_qualimap.zip" | |||
| } | |||
| } | |||
+ 39
- 0
tasks/quartet_mendelian.wdl
Прегледај датотеку
| @@ -0,0 +1,39 @@ | |||
| task quartet_mendelian { | |||
| Array[File] project_mendelian_summary | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| for i in ${sep=" " project_mendelian_summary} | |||
| do | |||
| cat $i | sed -n '2,3p' >> mendelian.summary | |||
| done | |||
| sed '1iFamily\tTotal_Variants\tMendelian_Concordant_Variants\tMendelian_Concordance_Rate' mendelian.summary > mendelian.txt | |||
| cat mendelian.txt | grep 'INDEL' | cut -f4 | grep -v 'Mendelian_Concordance_Rate' | awk '{for(i=1;i<=NF;i++) {sum[i] += $i; sumsq[i] += ($i)^2}} | |||
| END {for (i=1;i<=NF;i++) { | |||
| printf "%f %f \n", sum[i]/NR, sqrt((sumsq[i]-sum[i]^2/NR)/NR)} | |||
| }' >> quartet_indel_aver-std.txt | |||
| cat mendelian.txt | grep 'SNV' | cut -f4 | grep -v 'Mendelian_Concordance_Rate' | awk '{for(i=1;i<=NF;i++) {sum[i] += $i; sumsq[i] += ($i)^2}} | |||
| END {for (i=1;i<=NF;i++) { | |||
| printf "%f %f \n", sum[i]/NR, sqrt((sumsq[i]-sum[i]^2/NR)/NR)} | |||
| }' >> quartet_snv_aver-std.txt | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File mendelian_summary="mendelian.txt" | |||
| File snv_aver_std = "quartet_snv_aver-std.txt" | |||
| File indel_aver_std = "quartet_indel_aver-std.txt" | |||
| } | |||
| } | |||
+ 63
- 0
tasks/rename_fastq.wdl
Прегледај датотеку
| @@ -0,0 +1,63 @@ | |||
| task rename_fastq { | |||
| File fastq_1_D5 | |||
| String fastq_1_D5_name = sub(basename(fastq_1_D5, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| File fastq_1_D6 | |||
| String fastq_1_D6_name = sub(basename(fastq_1_D6, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| File fastq_1_F7 | |||
| String fastq_1_F7_name = sub(basename(fastq_1_F7, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| File fastq_1_M8 | |||
| String fastq_1_M8_name = sub(basename(fastq_1_M8, "_R1.fastq.gz"), "_R1.fq.gz$", "") | |||
| File fastq_2_D5 | |||
| String fastq_2_D5_name = sub(basename(fastq_2_D5, "_R2.fastq.gz"), "_R2.fq.gz$", "") | |||
| File fastq_2_D6 | |||
| String fastq_2_D6_name = sub(basename(fastq_2_D6, "_R2.fastq.gz"), "_R2.fq.gz$", "") | |||
| File fastq_2_F7 | |||
| String fastq_2_F7_name = sub(basename(fastq_2_F7, "_R2.fastq.gz"), "_R2.fq.gz$", "") | |||
| File fastq_2_M8 | |||
| String fastq_2_M8_name = sub(basename(fastq_2_M8, "_R2.fastq.gz"), "_R2.fq.gz$", "") | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cp ${fastq_1_D5} ${fastq_1_D5_name}_${project}_LCL5_R1.fastq.gz | |||
| rm ${fastq_1_D5} | |||
| cp ${fastq_1_D6} ${fastq_1_D6_name}_${project}_LCL6_R1.fastq.gz | |||
| rm ${fastq_1_D6} | |||
| cp ${fastq_1_F7} ${fastq_1_F7_name}_${project}_LCL7_R1.fastq.gz | |||
| rm ${fastq_1_F7} | |||
| cp ${fastq_1_M8} ${fastq_1_M8_name}_${project}_LCL8_R1.fastq.gz | |||
| rm ${fastq_1_M8} | |||
| cp ${fastq_2_D5} ${fastq_2_D5_name}_${project}_LCL5_R2.fastq.gz | |||
| rm ${fastq_2_D5} | |||
| cp ${fastq_2_D6} ${fastq_2_D6_name}_${project}_LCL6_R2.fastq.gz | |||
| rm ${fastq_2_D6} | |||
| cp ${fastq_2_F7} ${fastq_2_F7_name}_${project}_LCL7_R2.fastq.gz | |||
| rm ${fastq_2_F7} | |||
| cp ${fastq_2_M8} ${fastq_2_M8_name}_${project}_LCL8_R2.fastq.gz | |||
| rm ${fastq_2_M8} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File fastq_1_D5_renamed = "${fastq_1_D5_name}_${project}_LCL5_R1.fastq.gz" | |||
| File fastq_1_D6_renamed = "${fastq_1_D6_name}_${project}_LCL6_R1.fastq.gz" | |||
| File fastq_1_F7_renamed = "${fastq_1_F7_name}_${project}_LCL7_R1.fastq.gz" | |||
| File fastq_1_M8_renamed = "${fastq_1_M8_name}_${project}_LCL8_R1.fastq.gz" | |||
| File fastq_2_D5_renamed = "${fastq_2_D5_name}_${project}_LCL5_R2.fastq.gz" | |||
| File fastq_2_D6_renamed = "${fastq_2_D6_name}_${project}_LCL6_R2.fastq.gz" | |||
| File fastq_2_F7_renamed = "${fastq_2_F7_name}_${project}_LCL7_R2.fastq.gz" | |||
| File fastq_2_M8_renamed = "${fastq_2_M8_name}_${project}_LCL8_R2.fastq.gz" | |||
| } | |||
| } | |||
+ 41
- 0
tasks/rename_vcf.wdl
Прегледај датотеку
| @@ -0,0 +1,41 @@ | |||
| task rename_vcf { | |||
| File vcf_D5 | |||
| String vcf_D5_name = basename(vcf_D5, ".vcf") | |||
| File vcf_D6 | |||
| String vcf_D6_name = basename(vcf_D6, ".vcf") | |||
| File vcf_F7 | |||
| String vcf_F7_name = basename(vcf_F7, ".vcf") | |||
| File vcf_M8 | |||
| String vcf_M8_name = basename(vcf_M8, ".vcf") | |||
| String project | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| cp ${vcf_D5} ${vcf_D5_name}_${project}_LCL5.vcf | |||
| rm ${vcf_D5} | |||
| cp ${vcf_D6} ${vcf_D6_name}_${project}_LCL6.vcf | |||
| rm ${vcf_D6} | |||
| cp ${vcf_F7} ${vcf_F7_name}_${project}_LCL7.vcf | |||
| rm ${vcf_F7} | |||
| cp ${vcf_M8} ${vcf_M8_name}_${project}_LCL8.vcf | |||
| rm ${vcf_M8} | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File vcf_D5_renamed = "${vcf_D5_name}_${project}_LCL5.vcf" | |||
| File vcf_D6_renamed = "${vcf_D6_name}_${project}_LCL6.vcf" | |||
| File vcf_F7_renamed = "${vcf_F7_name}_${project}_LCL7.vcf" | |||
| File vcf_M8_renamed = "${vcf_M8_name}_${project}_LCL8.vcf" | |||
| } | |||
| } | |||
+ 54
- 0
tasks/sentieon.wdl
Прегледај датотеку
| @@ -0,0 +1,54 @@ | |||
| task sentieon { | |||
| File quality_yield | |||
| File wgs_metrics_algo | |||
| File aln_metrics | |||
| File is_metrics | |||
| File hs_metrics | |||
| String sample = basename(quality_yield,"_deduped_QualityYield.txt") | |||
| String docker | |||
| String cluster_config | |||
| String disk_size | |||
| command <<< | |||
| set -o pipefail | |||
| set -e | |||
| cat ${quality_yield} | sed -n '2,2p' > quality_yield.header | |||
| cat ${quality_yield} | sed -n '3,3p' > ${sample}.quality_yield | |||
| cat ${wgs_metrics_algo} | sed -n '2,2p' > wgs_metrics_algo.header | |||
| cat ${wgs_metrics_algo} | sed -n '3,3p' > ${sample}.wgs_metrics_algo | |||
| cat ${aln_metrics} | sed -n '2,2p' > aln_metrics.header | |||
| cat ${aln_metrics} | sed -n '5,5p' > ${sample}.aln_metrics | |||
| cat ${is_metrics} | sed -n '2,2p' > is_metrics.header | |||
| cat ${is_metrics} | sed -n '3,3p' > ${sample}.is_metrics | |||
| cat ${hs_metrics} | sed -n '2,2p' > hs_metrics.header | |||
| cat ${hs_metrics} | sed -n '3,3p' > ${sample}.hs_mtrics | |||
| >>> | |||
| runtime { | |||
| docker:docker | |||
| cluster:cluster_config | |||
| systemDisk:"cloud_ssd 40" | |||
| dataDisk:"cloud_ssd " + disk_size + " /cromwell_root/" | |||
| } | |||
| output { | |||
| File quality_yield_header = "quality_yield.header" | |||
| File quality_yield_data = "${sample}.quality_yield" | |||
| File wgs_metrics_algo_header = "wgs_metrics_algo.header" | |||
| File wgs_metrics_algo_data = "${sample}.wgs_metrics_algo" | |||
| File aln_metrics_header = "aln_metrics.header" | |||
| File aln_metrics_data = "${sample}.aln_metrics" | |||
| File is_metrics_header = "is_metrics.header" | |||
| File is_metrics_data = "${sample}.is_metrics" | |||
| File hs_metrics_header = "hs_metrics.header" | |||
| File hs_metrics_data = "${sample}.hs_metrics" | |||
| } | |||
| } | |||
+ 977
- 0
workflow.wdl
Прегледај датотеку
| @@ -0,0 +1,977 @@ | |||
| import "./tasks/rename_fastq.wdl" as rename_fastq | |||
| import "./tasks/rename_vcf.wdl" as rename_vcf | |||
| import "./tasks/mapping.wdl" as mapping | |||
| import "./tasks/Dedup.wdl" as Dedup | |||
| import "./tasks/qualimap.wdl" as qualimap | |||
| import "./tasks/deduped_Metrics.wdl" as deduped_Metrics | |||
| import "./tasks/sentieon.wdl" as sentieon | |||
| import "./tasks/Realigner.wdl" as Realigner | |||
| import "./tasks/BQSR.wdl" as BQSR | |||
| import "./tasks/Haplotyper.wdl" as Haplotyper | |||
| import "./tasks/benchmark.wdl" as benchmark | |||
| import "./tasks/multiqc.wdl" as multiqc | |||
| import "./tasks/multiqc_hap.wdl" as multiqc_hap | |||
| import "./tasks/merge_sentieon_metrics.wdl" as merge_sentieon_metrics | |||
| import "./tasks/extract_tables.wdl" as extract_tables | |||
| import "./tasks/extract_tables_vcf.wdl" as extract_tables_vcf | |||
| import "./tasks/mendelian.wdl" as mendelian | |||
| import "./tasks/merge_mendelian.wdl" as merge_mendelian | |||
| import "./tasks/quartet_mendelian.wdl" as quartet_mendelian | |||
| import "./tasks/fastqc.wdl" as fastqc | |||
| import "./tasks/fastqscreen.wdl" as fastqscreen | |||
| import "./tasks/merge_family.wdl" as merge_family | |||
| import "./tasks/filter_vcf.wdl" as filter_vcf | |||
| import "./tasks/bed_to_interval_list.wdl" as bed_to_interval_list | |||
| workflow {{ project_name }} { | |||
| File? fastq_1_D5 | |||
| File? fastq_1_D6 | |||
| File? fastq_1_F7 | |||
| File? fastq_1_M8 | |||
| File? fastq_2_D5 | |||
| File? fastq_2_D6 | |||
| File? fastq_2_F7 | |||
| File? fastq_2_M8 | |||
| File? vcf_D5 | |||
| File? vcf_D6 | |||
| File? vcf_F7 | |||
| File? vcf_M8 | |||
| File bed | |||
| String SENTIEON_INSTALL_DIR | |||
| String SENTIEON_LICENSE | |||
| String SENTIEONdocker | |||
| String FASTQCdocker | |||
| String FASTQSCREENdocker | |||
| String QUALIMAPdocker | |||
| String BENCHMARKdocker | |||
| String MENDELIANdocker | |||
| String DIYdocker | |||
| String MULTIQCdocker | |||
| String BEDTOOLSdocker | |||
| String PICARDdocker | |||
| String fasta | |||
| File ref_dir | |||
| File dbmills_dir | |||
| String db_mills | |||
| File dbsnp_dir | |||
| String dbsnp | |||
| String pl | |||
| File reference_bed_dict | |||
| File screen_ref_dir | |||
| File fastq_screen_conf | |||
| File benchmarking_dir | |||
| File benchmark_region | |||
| String project | |||
| String disk_size | |||
| String BIGcluster_config | |||
| String SMALLcluster_config | |||
| if (fastq_1_D5!= "") { | |||
| call bed_to_interval_list.bed_to_interval_list as bed_to_interval_list { | |||
| input: | |||
| bed=bed, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| interval_list_name=project, | |||
| reference_bed_dict=reference_bed_dict, | |||
| docker=PICARDdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mapping.mapping as mapping_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| SENTIEON_LICENSE=SENTIEON_LICENSE, | |||
| pl=pl, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| fastq_1=fastq_1_D5, | |||
| fastq_2=fastq_2_D5, | |||
| group=project, | |||
| sample='LCL5', | |||
| project=project, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call fastqc.fastqc as fastqc_D5 { | |||
| input: | |||
| read1=fastq_1_D5, | |||
| read2=fastq_2_D5, | |||
| project=project, | |||
| sample="LCL5", | |||
| docker=FASTQCdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen_D5 { | |||
| input: | |||
| read1=fastq_1_D5, | |||
| read2=fastq_2_D5, | |||
| project=project, | |||
| sample="LCL5", | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf, | |||
| docker=FASTQSCREENdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Dedup.Dedup as Dedup_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=mapping_D5.sorted_bam, | |||
| sorted_bam_index=mapping_D5.sorted_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call qualimap.qualimap as qualimap_D5 { | |||
| input: | |||
| bam=Dedup_D5.Dedup_bam, | |||
| bai=Dedup_D5.Dedup_bam_index, | |||
| bed=bed, | |||
| docker=QUALIMAPdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as deduped_Metrics_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| bed=bed, | |||
| Dedup_bam=Dedup_D5.Dedup_bam, | |||
| Dedup_bam_index=Dedup_D5.Dedup_bam_index, | |||
| docker=SENTIEONdocker, | |||
| interval_list=bed_to_interval_list.interval_list, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call sentieon.sentieon as sentieon_D5 { | |||
| input: | |||
| quality_yield=deduped_Metrics_D5.deduped_QualityYield, | |||
| wgs_metrics_algo=deduped_Metrics_D5.deduped_wgsmetrics, | |||
| aln_metrics=deduped_Metrics_D5.dedeuped_aln_metrics, | |||
| is_metrics=deduped_Metrics_D5.deduped_is_metrics, | |||
| hs_metrics=deduped_Metrics_D5.deduped_hsmetrics, | |||
| docker=SENTIEONdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Realigner.Realigner as Realigner_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=Dedup_D5.Dedup_bam, | |||
| Dedup_bam_index=Dedup_D5.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call BQSR.BQSR as BQSR_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| realigned_bam=Realigner_D5.realigner_bam, | |||
| realigned_bam_index=Realigner_D5.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call Haplotyper.Haplotyper as Haplotyper_D5 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=BQSR_D5.recaled_bam, | |||
| recaled_bam_index=BQSR_D5.recaled_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_D5 { | |||
| input: | |||
| vcf=Haplotyper_D5.vcf, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_D5 { | |||
| input: | |||
| filtered_vcf=filter_vcf_D5.filtered_vcf, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| bed=filter_vcf_D5.filtered_bed, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mapping.mapping as mapping_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| SENTIEON_LICENSE=SENTIEON_LICENSE, | |||
| pl=pl, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| fastq_1=fastq_1_D6, | |||
| fastq_2=fastq_2_D6, | |||
| group=project, | |||
| sample='LCL6', | |||
| project=project, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call fastqc.fastqc as fastqc_D6 { | |||
| input: | |||
| read1=fastq_1_D6, | |||
| read2=fastq_2_D6, | |||
| project=project, | |||
| sample="LCL6", | |||
| docker=FASTQCdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen_D6 { | |||
| input: | |||
| read1=fastq_1_D6, | |||
| read2=fastq_2_D6, | |||
| project=project, | |||
| sample="LCL6", | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf, | |||
| docker=FASTQSCREENdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Dedup.Dedup as Dedup_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=mapping_D6.sorted_bam, | |||
| sorted_bam_index=mapping_D6.sorted_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call qualimap.qualimap as qualimap_D6 { | |||
| input: | |||
| bam=Dedup_D6.Dedup_bam, | |||
| bai=Dedup_D6.Dedup_bam_index, | |||
| bed=bed, | |||
| docker=QUALIMAPdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as deduped_Metrics_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| bed=bed, | |||
| Dedup_bam=Dedup_D6.Dedup_bam, | |||
| Dedup_bam_index=Dedup_D6.Dedup_bam_index, | |||
| interval_list=bed_to_interval_list.interval_list, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call sentieon.sentieon as sentieon_D6 { | |||
| input: | |||
| quality_yield=deduped_Metrics_D6.deduped_QualityYield, | |||
| wgs_metrics_algo=deduped_Metrics_D6.deduped_wgsmetrics, | |||
| aln_metrics=deduped_Metrics_D6.dedeuped_aln_metrics, | |||
| is_metrics=deduped_Metrics_D6.deduped_is_metrics, | |||
| hs_metrics=deduped_Metrics_D6.deduped_hsmetrics, | |||
| docker=SENTIEONdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Realigner.Realigner as Realigner_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=Dedup_D6.Dedup_bam, | |||
| Dedup_bam_index=Dedup_D6.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call BQSR.BQSR as BQSR_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| realigned_bam=Realigner_D6.realigner_bam, | |||
| realigned_bam_index=Realigner_D6.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call Haplotyper.Haplotyper as Haplotyper_D6 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=BQSR_D6.recaled_bam, | |||
| recaled_bam_index=BQSR_D6.recaled_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_D6 { | |||
| input: | |||
| vcf=Haplotyper_D6.vcf, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_D6 { | |||
| input: | |||
| filtered_vcf=filter_vcf_D6.filtered_vcf, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| bed=filter_vcf_D6.filtered_bed, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mapping.mapping as mapping_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| SENTIEON_LICENSE=SENTIEON_LICENSE, | |||
| pl=pl, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| fastq_1=fastq_1_F7, | |||
| fastq_2=fastq_2_F7, | |||
| group=project, | |||
| sample='LCL7', | |||
| project=project, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call fastqc.fastqc as fastqc_F7 { | |||
| input: | |||
| read1=fastq_1_F7, | |||
| read2=fastq_2_F7, | |||
| project=project, | |||
| sample="LCL7", | |||
| docker=FASTQCdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen_F7 { | |||
| input: | |||
| read1=fastq_1_F7, | |||
| read2=fastq_2_F7, | |||
| project=project, | |||
| sample="LCL7", | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf, | |||
| docker=FASTQSCREENdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Dedup.Dedup as Dedup_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=mapping_F7.sorted_bam, | |||
| sorted_bam_index=mapping_F7.sorted_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call qualimap.qualimap as qualimap_F7 { | |||
| input: | |||
| bam=Dedup_F7.Dedup_bam, | |||
| bai=Dedup_F7.Dedup_bam_index, | |||
| bed=bed, | |||
| docker=QUALIMAPdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as deduped_Metrics_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| bed=bed, | |||
| Dedup_bam=Dedup_F7.Dedup_bam, | |||
| Dedup_bam_index=Dedup_F7.Dedup_bam_index, | |||
| interval_list=bed_to_interval_list.interval_list, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call sentieon.sentieon as sentieon_F7 { | |||
| input: | |||
| quality_yield=deduped_Metrics_F7.deduped_QualityYield, | |||
| wgs_metrics_algo=deduped_Metrics_F7.deduped_wgsmetrics, | |||
| aln_metrics=deduped_Metrics_F7.dedeuped_aln_metrics, | |||
| is_metrics=deduped_Metrics_F7.deduped_is_metrics, | |||
| hs_metrics=deduped_Metrics_F7.deduped_hsmetrics, | |||
| docker=SENTIEONdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Realigner.Realigner as Realigner_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=Dedup_F7.Dedup_bam, | |||
| Dedup_bam_index=Dedup_F7.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call BQSR.BQSR as BQSR_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| realigned_bam=Realigner_F7.realigner_bam, | |||
| realigned_bam_index=Realigner_F7.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call Haplotyper.Haplotyper as Haplotyper_F7 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=BQSR_F7.recaled_bam, | |||
| recaled_bam_index=BQSR_F7.recaled_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_F7 { | |||
| input: | |||
| vcf=Haplotyper_F7.vcf, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| project=project, | |||
| benchmark_region=benchmark_region, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_F7 { | |||
| input: | |||
| filtered_vcf=filter_vcf_F7.filtered_vcf, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| bed=filter_vcf_F7.filtered_bed, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call mapping.mapping as mapping_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| SENTIEON_LICENSE=SENTIEON_LICENSE, | |||
| pl=pl, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| fastq_1=fastq_1_M8, | |||
| fastq_2=fastq_2_M8, | |||
| group=project, | |||
| project=project, | |||
| sample='LCL8', | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call fastqc.fastqc as fastqc_M8 { | |||
| input: | |||
| read1=fastq_1_M8, | |||
| read2=fastq_2_M8, | |||
| project=project, | |||
| sample="LCL8", | |||
| docker=FASTQCdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call fastqscreen.fastq_screen as fastqscreen_M8 { | |||
| input: | |||
| read1=fastq_1_M8, | |||
| read2=fastq_2_M8, | |||
| project=project, | |||
| sample="LCL8", | |||
| screen_ref_dir=screen_ref_dir, | |||
| fastq_screen_conf=fastq_screen_conf, | |||
| docker=FASTQSCREENdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Dedup.Dedup as Dedup_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| sorted_bam=mapping_M8.sorted_bam, | |||
| sorted_bam_index=mapping_M8.sorted_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call qualimap.qualimap as qualimap_M8 { | |||
| input: | |||
| bam=Dedup_M8.Dedup_bam, | |||
| bai=Dedup_M8.Dedup_bam_index, | |||
| docker=QUALIMAPdocker, | |||
| bed=bed, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call deduped_Metrics.deduped_Metrics as deduped_Metrics_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| bed=bed, | |||
| Dedup_bam=Dedup_M8.Dedup_bam, | |||
| Dedup_bam_index=Dedup_M8.Dedup_bam_index, | |||
| interval_list=bed_to_interval_list.interval_list, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call sentieon.sentieon as sentieon_M8 { | |||
| input: | |||
| quality_yield=deduped_Metrics_M8.deduped_QualityYield, | |||
| wgs_metrics_algo=deduped_Metrics_M8.deduped_wgsmetrics, | |||
| aln_metrics=deduped_Metrics_M8.dedeuped_aln_metrics, | |||
| is_metrics=deduped_Metrics_M8.deduped_is_metrics, | |||
| hs_metrics=deduped_Metrics_M8.deduped_hsmetrics, | |||
| docker=SENTIEONdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call Realigner.Realigner as Realigner_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| Dedup_bam=Dedup_M8.Dedup_bam, | |||
| Dedup_bam_index=Dedup_M8.Dedup_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call BQSR.BQSR as BQSR_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| realigned_bam=Realigner_M8.realigner_bam, | |||
| realigned_bam_index=Realigner_M8.realigner_bam_index, | |||
| db_mills=db_mills, | |||
| dbmills_dir=dbmills_dir, | |||
| dbsnp=dbsnp, | |||
| dbsnp_dir=dbsnp_dir, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call Haplotyper.Haplotyper as Haplotyper_M8 { | |||
| input: | |||
| SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, | |||
| fasta=fasta, | |||
| ref_dir=ref_dir, | |||
| recaled_bam=BQSR_M8.recaled_bam, | |||
| recaled_bam_index=BQSR_M8.recaled_bam_index, | |||
| docker=SENTIEONdocker, | |||
| disk_size=disk_size, | |||
| cluster_config=BIGcluster_config | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_M8 { | |||
| input: | |||
| vcf=Haplotyper_M8.vcf, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| project=project, | |||
| benchmark_region=benchmark_region, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_M8 { | |||
| input: | |||
| filtered_vcf=filter_vcf_M8.filtered_vcf, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| bed=filter_vcf_M8.filtered_bed, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| Array[File] fastqc_read1_zip = [fastqc_D5.read1_zip, fastqc_D6.read1_zip, fastqc_F7.read1_zip, fastqc_M8.read1_zip] | |||
| Array[File] fastqc_read2_zip = [fastqc_D5.read2_zip, fastqc_D6.read2_zip, fastqc_F7.read2_zip, fastqc_M8.read2_zip] | |||
| Array[File] fastqscreen_txt1 = [fastqscreen_D5.txt1, fastqscreen_D6.txt1, fastqscreen_F7.txt1, fastqscreen_M8.txt1] | |||
| Array[File] fastqscreen_txt2 = [fastqscreen_D5.txt2, fastqscreen_D6.txt2, fastqscreen_F7.txt2, fastqscreen_M8.txt2] | |||
| Array[File] benchmark_summary = [benchmark_D5.summary, benchmark_D6.summary, benchmark_F7.summary, benchmark_M8.summary] | |||
| call multiqc.multiqc as multiqc_big { | |||
| input: | |||
| read1_zip=fastqc_read1_zip, | |||
| read2_zip=fastqc_read2_zip, | |||
| txt1=fastqscreen_txt1, | |||
| txt2=fastqscreen_txt2, | |||
| summary=benchmark_summary, | |||
| docker=MULTIQCdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| Array[File] sentieon_quality_yield_header = [sentieon_D5.quality_yield_header, sentieon_D6.quality_yield_header, sentieon_F7.quality_yield_header, sentieon_M8.quality_yield_header] | |||
| Array[File] sentieon_wgs_metrics_algo_header = [sentieon_D5.wgs_metrics_algo_header, sentieon_D6.wgs_metrics_algo_header, sentieon_F7.wgs_metrics_algo_header, sentieon_M8.wgs_metrics_algo_header] | |||
| Array[File] sentieon_aln_metrics_header = [sentieon_D5.aln_metrics_header, sentieon_D6.aln_metrics_header, sentieon_F7.aln_metrics_header, sentieon_M8.aln_metrics_header] | |||
| Array[File] sentieon_is_metrics_header = [sentieon_D5.is_metrics_header, sentieon_D6.is_metrics_header, sentieon_F7.is_metrics_header, sentieon_M8.is_metrics_header] | |||
| Array[File] sentieon_hs_metrics_header = [sentieon_D5.hs_metrics_header, sentieon_D6.hs_metrics_header, sentieon_F7.hs_metrics_header, sentieon_M8.hs_metrics_header] | |||
| Array[File] sentieon_quality_yield_data = [sentieon_D5.quality_yield_data, sentieon_D6.quality_yield_data, sentieon_F7.quality_yield_data, sentieon_M8.quality_yield_data] | |||
| Array[File] sentieon_wgs_metrics_algo_data = [sentieon_D5.wgs_metrics_algo_data, sentieon_D6.wgs_metrics_algo_data, sentieon_F7.wgs_metrics_algo_data, sentieon_M8.wgs_metrics_algo_data] | |||
| Array[File] sentieon_aln_metrics_data = [sentieon_D5.aln_metrics_data, sentieon_D6.aln_metrics_data, sentieon_F7.aln_metrics_data, sentieon_M8.aln_metrics_data] | |||
| Array[File] sentieon_is_metrics_data = [sentieon_D5.is_metrics_data, sentieon_D6.is_metrics_data, sentieon_F7.is_metrics_data, sentieon_M8.is_metrics_data] | |||
| Array[File] sentieon_hs_metrics_data = [sentieon_D5.hs_metrics_data, sentieon_D6.hs_metrics_data, sentieon_F7.hs_metrics_data, sentieon_M8.hs_metrics_data] | |||
| call merge_sentieon_metrics.merge_sentieon_metrics as merge_sentieon_metrics { | |||
| input: | |||
| quality_yield_header=sentieon_quality_yield_header, | |||
| wgs_metrics_algo_header=sentieon_wgs_metrics_algo_header, | |||
| aln_metrics_header=sentieon_aln_metrics_header, | |||
| is_metrics_header=sentieon_is_metrics_header, | |||
| hs_metrics_header=sentieon_hs_metrics_header, | |||
| quality_yield_data=sentieon_quality_yield_data, | |||
| wgs_metrics_algo_data=sentieon_wgs_metrics_algo_data, | |||
| aln_metrics_data=sentieon_aln_metrics_data, | |||
| is_metrics_data=sentieon_is_metrics_data, | |||
| hs_metrics_data=sentieon_hs_metrics_data, | |||
| project=project, | |||
| docker=MULTIQCdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call extract_tables.extract_tables as extract_tables { | |||
| input: | |||
| quality_yield_summary=merge_sentieon_metrics.quality_yield_summary, | |||
| wgs_metrics_summary=merge_sentieon_metrics.wgs_metrics_summary, | |||
| aln_metrics_summary=merge_sentieon_metrics.aln_metrics_summary, | |||
| is_metrics_summary=merge_sentieon_metrics.is_metrics_summary, | |||
| hs_metrics_summary=merge_sentieon_metrics.hs_metrics_summary, | |||
| fastqc=multiqc_big.fastqc, | |||
| fastqscreen=multiqc_big.fastqscreen, | |||
| hap=multiqc_big.hap, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call merge_family.merge_family as merge_family { | |||
| input: | |||
| D5_vcf=benchmark_D5.rtg_vcf, | |||
| D6_vcf=benchmark_D6.rtg_vcf, | |||
| F7_vcf=benchmark_F7.rtg_vcf, | |||
| M8_vcf=benchmark_M8.rtg_vcf, | |||
| D5_vcf_tbi=benchmark_D5.rtg_vcf_index, | |||
| D6_vcf_tbi=benchmark_D6.rtg_vcf_index, | |||
| F7_vcf_tbi=benchmark_F7.rtg_vcf_index, | |||
| M8_vcf_tbi=benchmark_M8.rtg_vcf_index, | |||
| project=project, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size, | |||
| } | |||
| call mendelian.mendelian as mendelian { | |||
| input: | |||
| family_vcf=merge_family.family_vcf, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=MENDELIANdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call merge_mendelian.merge_mendelian as merge_mendelian { | |||
| input: | |||
| D5_trio_vcf=mendelian.D5_trio_vcf, | |||
| D6_trio_vcf=mendelian.D6_trio_vcf, | |||
| family_vcf=merge_family.family_vcf, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| if (vcf_D5!= "") { | |||
| call rename_vcf.rename_vcf as rename_vcf{ | |||
| input: | |||
| vcf_D5=vcf_D5, | |||
| vcf_D6=vcf_D6, | |||
| vcf_F7=vcf_F7, | |||
| vcf_M8=vcf_M8, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_D5_vcf { | |||
| input: | |||
| vcf=rename_vcf.vcf_D5_renamed, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| docker=BEDTOOLSdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_D5_vcf { | |||
| input: | |||
| filtered_vcf=filter_vcf_D5_vcf.filtered_vcf, | |||
| bed=filter_vcf_D5_vcf.filtered_bed, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_D6_vcf { | |||
| input: | |||
| vcf=rename_vcf.vcf_D6_renamed, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_D6_vcf { | |||
| input: | |||
| filtered_vcf=filter_vcf_D6_vcf.filtered_vcf, | |||
| bed=filter_vcf_D6_vcf.filtered_bed, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_F7_vcf { | |||
| input: | |||
| vcf=rename_vcf.vcf_F7_renamed, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_F7_vcf { | |||
| input: | |||
| filtered_vcf=filter_vcf_F7_vcf.filtered_vcf, | |||
| bed=filter_vcf_F7_vcf.filtered_bed, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call filter_vcf.filter_vcf as filter_vcf_M8_vcf { | |||
| input: | |||
| vcf=rename_vcf.vcf_M8_renamed, | |||
| docker=BEDTOOLSdocker, | |||
| bed=bed, | |||
| benchmark_region=benchmark_region, | |||
| project=project, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call benchmark.benchmark as benchmark_M8_vcf { | |||
| input: | |||
| filtered_vcf=filter_vcf_M8_vcf.filtered_vcf, | |||
| bed=filter_vcf_M8_vcf.filtered_bed, | |||
| benchmarking_dir=benchmarking_dir, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| Array[File] benchmark_summary_hap = [benchmark_D5_vcf.summary, benchmark_D6_vcf.summary, benchmark_F7_vcf.summary, benchmark_M8_vcf.summary] | |||
| call multiqc_hap.multiqc_hap as multiqc_hap { | |||
| input: | |||
| summary=benchmark_summary_hap, | |||
| docker=MULTIQCdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call extract_tables_vcf.extract_tables_vcf as extract_tables_vcf { | |||
| input: | |||
| hap=multiqc_hap.hap, | |||
| project=project, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call merge_family.merge_family as merge_family_vcf { | |||
| input: | |||
| D5_vcf=benchmark_D5_vcf.rtg_vcf, | |||
| D6_vcf=benchmark_D6_vcf.rtg_vcf, | |||
| F7_vcf=benchmark_F7_vcf.rtg_vcf, | |||
| M8_vcf=benchmark_M8_vcf.rtg_vcf, | |||
| D5_vcf_tbi=benchmark_D5_vcf.rtg_vcf_index, | |||
| D6_vcf_tbi=benchmark_D6_vcf.rtg_vcf_index, | |||
| F7_vcf_tbi=benchmark_F7_vcf.rtg_vcf_index, | |||
| M8_vcf_tbi=benchmark_M8_vcf.rtg_vcf_index, | |||
| project=project, | |||
| docker=BENCHMARKdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size, | |||
| } | |||
| call mendelian.mendelian as mendelian_vcf { | |||
| input: | |||
| family_vcf=merge_family_vcf.family_vcf, | |||
| ref_dir=ref_dir, | |||
| fasta=fasta, | |||
| docker=MENDELIANdocker, | |||
| cluster_config=BIGcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| call merge_mendelian.merge_mendelian as merge_mendelian_vcf { | |||
| input: | |||
| D5_trio_vcf=mendelian_vcf.D5_trio_vcf, | |||
| D6_trio_vcf=mendelian_vcf.D6_trio_vcf, | |||
| family_vcf=merge_family_vcf.family_vcf, | |||
| docker=DIYdocker, | |||
| cluster_config=SMALLcluster_config, | |||
| disk_size=disk_size | |||
| } | |||
| } | |||
| } | |||
Loading…