
 LUYAO REN
633295b0f8
first commit
LUYAO REN
633295b0f8
first commit
|
4年前 | |
|---|---|---|
| picture | 4年前 | |
| tasks | 4年前 | |
| README.md | 4年前 | |
| defaults | 4年前 | |
| inputs | 4年前 | |
| workflow.wdl | 4年前 | |
README.md
NGS Check mates
Author: Ren Luyao
E-mail: 18110700050@fudan.edu.cn
Git: http://choppy.3steps.cn/renluyao/NGScheckMates.git
Last Updates: 2019/02/08
source activate choppy
choppy install renluyao/NGScheckMates
APP概述
NGScheckMates是用来检测某几个测序数据是否来自于同一个人,有以下几种应用场景:
(1)多组学研究,RNAseq和DNAseq是否是来自同一个人;
(2)被标注为配对的Tumor和Normal样本是否是来自同一个人;
(3)同一批样本,多次测序,其中有没有被标错的样本。
推荐直接用fastq模式,优点:如果有多种不同测序文件,比如一个项目中有WES和RNAseq,你要研究WES找到的候选突变是否影响了基因表达量的改变,你需要检查WES和RNAseq的数据是否来自同一个人,以确保分析结果的正确性。直接用fastq模式可以不用单独对RNAseq call germline mutation,以节省时间。这一步将单独运行一个脚本。
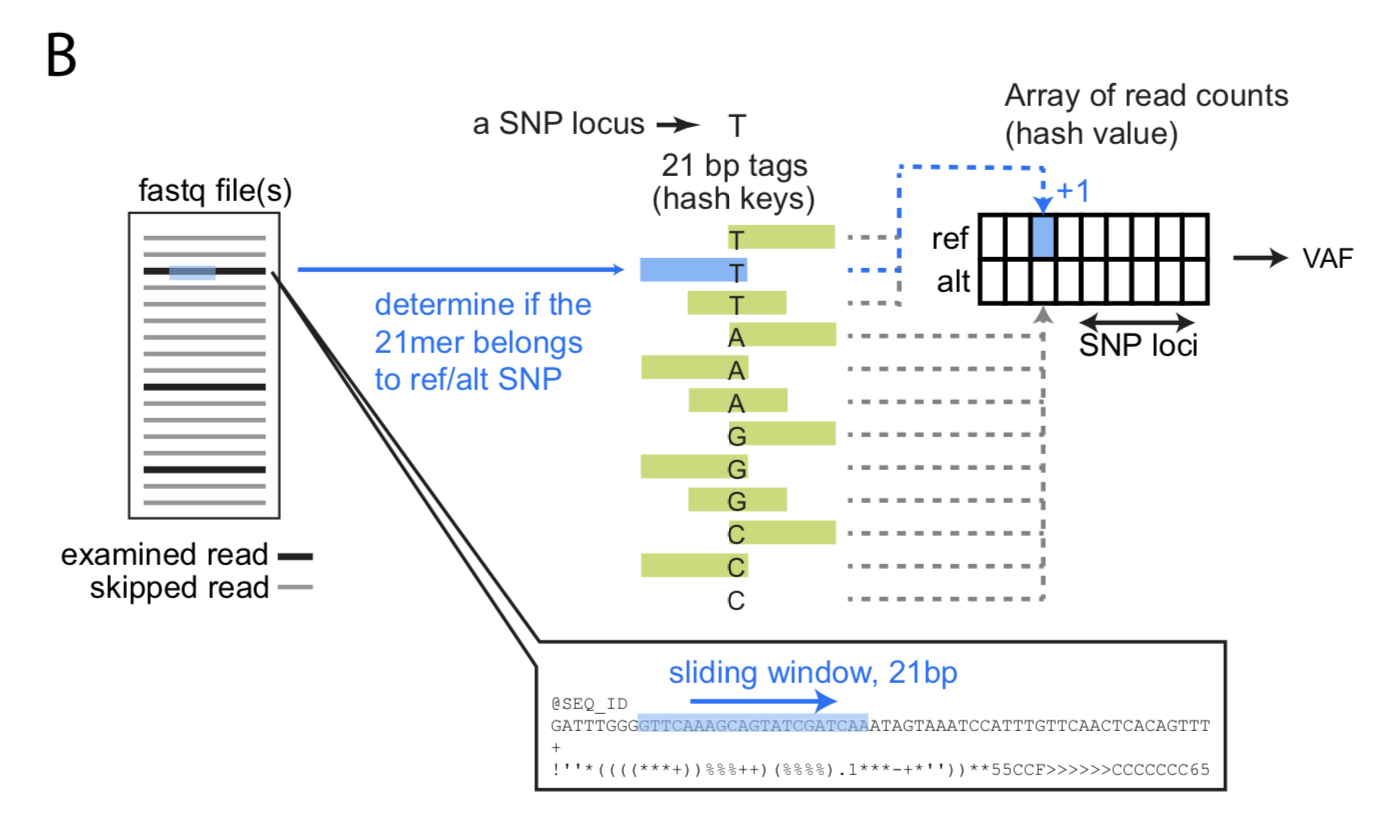
基于fastq文件检查样本的配对情况的原理是:他们首先从dbSNP数据库中选择了21067个位于外显子上的SNP用于预测样本配对。对于不用比对的fastq模式,他们在参考基因组中寻找了可以与参考基因组完全匹配的21bp长度的k-mer,位于这些k-mer上的SNP只剩下了11696个。然后用k-mer扫描fastq文件,计算每个SNP的VAF,再根据多个SNP的VAF计算样本间两两的相关性判断两个样本是否来源于一个人。
流程与参数
- Required arguments
-l 需要检测的fastq或者fastq.gz文件的表格,格式如下:
FASTQ_FILE1 (tab) FASTQ_FILE2 (tab) SAMPLE_NAME (\n)
Example:
/data/LSJ_R1.fastq /data/LSJ_R2.fastq LSJ
/data/LSH_R1.fastq /data/LSH_R2.fastq LSH
-pt 是一个包含SNP位点的二进制文件,这些位点可以用与样本的配对检查,在下载包中,路径为SNP/SNP.pt
-O 输出文件夹
- Optional arguments
-N 输出文件夹的前缀,default:“output”
-f 当你的样本中有父母与孩子或者兄弟姐妹时,加上这个参数,使用更严格的VAF相关系数的阈值
-nz Use the mean of non-zero depths across the SNPs as reference depth, default: Use the mean depth across all the SNPs
-s The read subsampling rate, default: 1.0
-d The target depth for read subsampling. NGSCheckMate calculates a subsampling rate based on this target depth.
-R The length of the genomic region with read mapping (default: 3x10^9) used to compute subsampling rate. If your data is NOT human WGS and you use the -d option, it is highly recommended that specify this value. For instance, if your data is human RNA-seq, the genomic length with read mapping is ~3% of the human genome (1x10^8)
注意:如果你的fastq文件特别大,它的计算会特别慢,三个参数的使用可以通过subsampling的方法加快运算速度,文献中报道只要0.5X深度的数据就能有很好的预测效果,有两种选择:
- 只使用 -s ,意思是fastq原始文件的百分之多少,如果你的fastq文件太大,运算速度很比较慢,可以使用其中一部分数据运算,不会影响运算结果,比如,30%就是
-s 0.3 - 需要通过使用-d 和-R
-L The length of the flanking sequence of the SNPs, default: 21bp. It is not recommended that you change this value unless you create your own pattern file (.pt) with a different length.
-p 线程数,default:1
APP输入变量与输入文件
(1)准备样本文件
choppy samples NGScheckMates --output samples
samples文件中输入是
- fastq_dir
fastq文件的地址,阿里云上的地址;如果需要使用多个项目的fastq文件,输入两个项目文件夹的上一级共同目录
- Input_file
一个txt文件,需要进行计算的文件的详细文件名,文件的地址按照要求修改
#read1 #read2 #sample_name
/cromwell_inputs/*/directory_name/read1.fastq.gz /cromwell_inputs/*/directory_name/read2.fastq.gz sample_name