
 LUYAO REN
6 年前
LUYAO REN
6 年前
共有 2 个文件被更改,包括 77 次插入 和 0 次删除
+ 77
- 0
README.md
查看文件
| @@ -0,0 +1,77 @@ | |||
| # BWA_FreeBayes | |||
| FreeBayes is a [Bayesian](http://en.wikipedia.org/wiki/Bayesian_inference) genetic variant detector designed to find small polymorphisms, specifically SNPs, indels, MNPs, and complex events (composite insertion and substitution events) smaller than the length of a short-read sequencing alignment. | |||
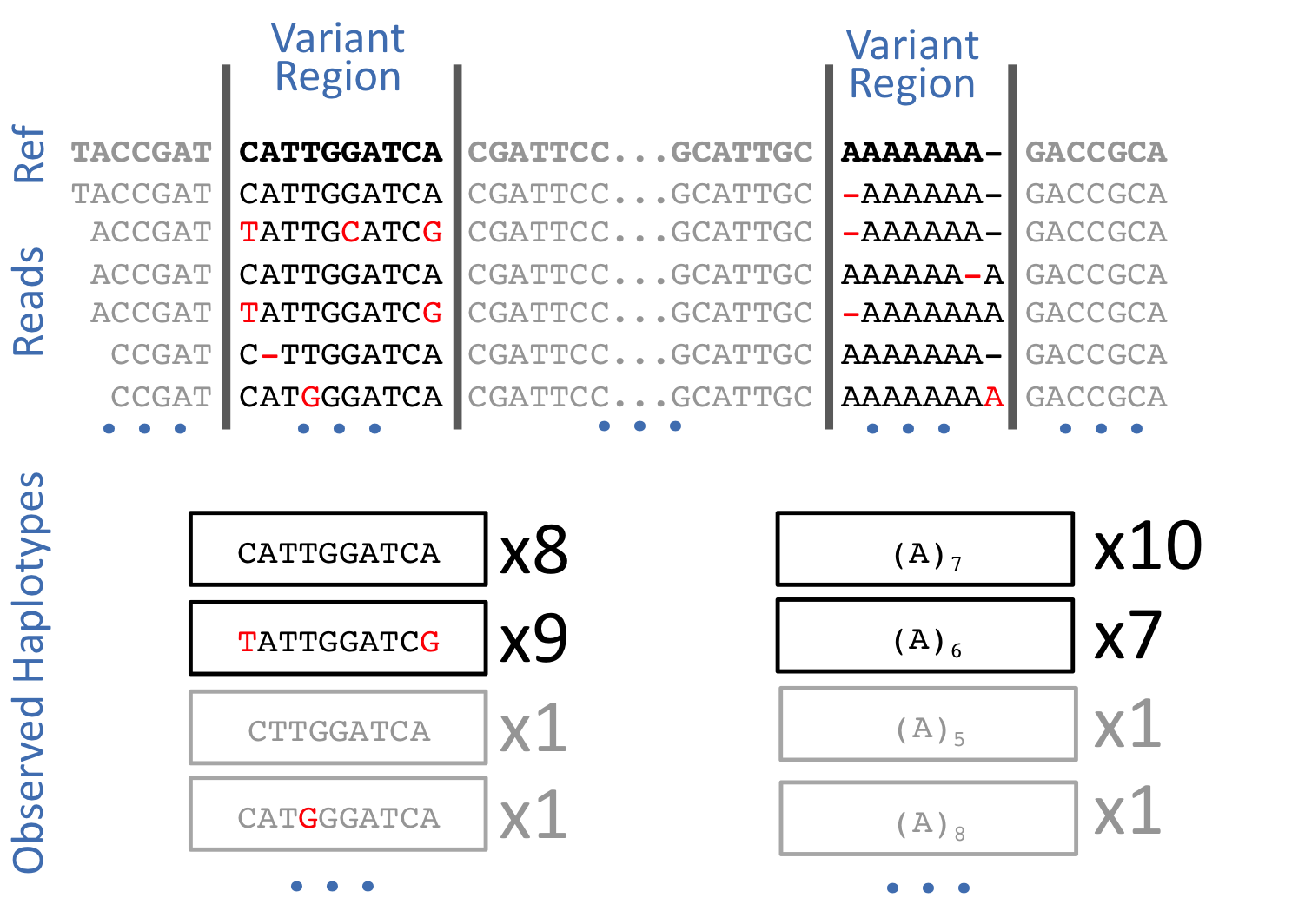
| FreeBayes is haplotype-based, in the sense that it calls variants based on the literal sequences of reads aligned to a particular target, not their precise alignment. This model is a straightforward generalization of previous ones (e.g. PolyBayes, samtools, GATK) which detect or report variants based on alignments. This method avoids one of the core problems with alignment-based variant detection--- that identical sequences may have multiple possible alignments: | |||
|  | |||
| FreeBayes uses short-read alignments for any number of individuals from a population and a reference genome to determine the most-likely combination of genotypes for the population at each position in the reference. It reports positions which it finds putatively polymorphic in variant call file format. It can also use an input set of variants (VCF) as a source of prior information, and a copy number variant map (BED) to define non-uniform ploidy variation across the samples under analysis. [1] | |||
| This pipeline uses BWA-MEM mapper from Sentieon and caller Freebayes. | |||
| ####**Freebayes default setting:** | |||
| `-C` variants supported by at least 2 observations in a single sample | |||
| `-F` and also at least 20% of the reads from a single sample | |||
| `--max-complex-gap` FreeBayes is capable of calling variant haplotypes shorter than a read length where multiple polymorphisms segregate on the same read. This parameter determines the maximum distance between polymorphisms phased in this way, which defaults to 3bp. In practice, this can comfortably be set to **half the read length**. | |||
| `--min-alternate-count` Require that **2** reads in one sample support an allele in order to consider it | |||
| `--min-alternate-fraction` or that the allele fraction in one sample is **0.2** | |||
| ####**Best practices and design philosophy** | |||
| FreeBayes incorporates a number of features in order to reduce the complexity of variant detection for researchers and developers: | |||
| - **Indel realignment is accomplished internally** using a read-independent method, and issues resulting from discordant alignments are dramatically reducedy through the direct detection of haplotypes. | |||
| - The need for **base quality recalibration is avoided** through the direct detection of haplotypes. Sequencing platform errors tend to cluster (e.g. at the ends of reads), and generate unique, non-repeating haplotypes at a given locus. | |||
| - **Variant quality recalibration is avoided** by incorporating a number of metrics, such as read placement bias and allele balance, directly into the Bayesian model. | |||
| So we use Dedup.bam from Sentieon, without doing indel realignment and BQSR. | |||
| ####**NIST's settings:** | |||
| `-F` 0.05, means at least 5% of the reads from a single sample support the variants | |||
| `-m` ,`--min-mapping-quality` 0, Exclude alignments from analysis if they have a mapping quality less than 1 (default). A mapping quality of zero means that the read maps to multiple locations with the same quality and that the mapper has picked one of these positions at random. | |||
| `--genotype-qualities` Calculate the marginal probability of genotypes and report as GQ in each sample field in the VCF output. | |||
| For now, I am not quite sure why they set min mapping quality to 0, so I will use default settings. | |||
| Other settings you can find by : | |||
| ``` | |||
| freebayes --help | |||
| ``` | |||
| Basic usage: | |||
| ```bash | |||
| freebayes -f ref.fa aln.bam >var.vcf | |||
| ``` | |||
| Command line used in this APP: | |||
| FreeBayes is very slow with single thread, using the scripts/freebayes-parallele script. | |||
| ```bash | |||
| freebayes-parallel <(fasta_generate_regions.py ref.fa.fai 100000) 36 \ | |||
| --genotype-qualities -f ref.fa aln.bam >var.vcf | |||
| ``` | |||
| This pipeline is for Quartet, if you have bam files, please refer to <http://choppy.3steps.cn/renluyao/FreeBayes> | |||
| ####**Reference** | |||
| 1. Freebayes GitHub <https://github.com/ekg/freebayes> | |||
| 2. Freebayes paper <https://arxiv.org/abs/1207.3907> | |||
二进制
pictures/haplotype_calling.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|
|
|
| 宽度: 1502 | 高度: 1064 | 大小: 171KB |
正在加载...