|
|
|
|
|
|
|
|
## minfi分析Illumina 850K(EPIC)
|
|
|
|
|
|
|
|
|
#Illumina 850K(EPIC)分析APP
|
|
|
|
|
|
|
|
|
#APP介绍
|
|
|
|
|
|
|
|
|
## 安装指南
|
|
|
|
|
|
|
|
|
###甲基化原理简述
|
|
|
|
|
|
|
|
|
`#激活choppy环境`
|
|
|
|
|
|
|
|
|
|
|
|
`'#sourse activate choppy'`
|
|
|
|
|
|
|
|
|
|
|
|
`#安装APP`
|
|
|
|
|
|
|
|
|
|
|
|
`choppy install XXXX`
|
|
|
|
|
|
|
|
|
|
|
|
### APP介绍
|
|
|
|
|
|
|
|
|
|
|
|
#### 甲基化原理简述
|
|
|
|
|
|
|
|
|
Illumina 850K甲基化芯片可同时检测>850,000个位点,覆盖>95%的CpG岛,99%的RefSeq基因,已经成为精准医学研究的重要方法之一。
|
|
|
Illumina 850K甲基化芯片可同时检测>850,000个位点,覆盖>95%的CpG岛,99%的RefSeq基因,已经成为精准医学研究的重要方法之一。
|
|
|
|
|
|
|
|
|
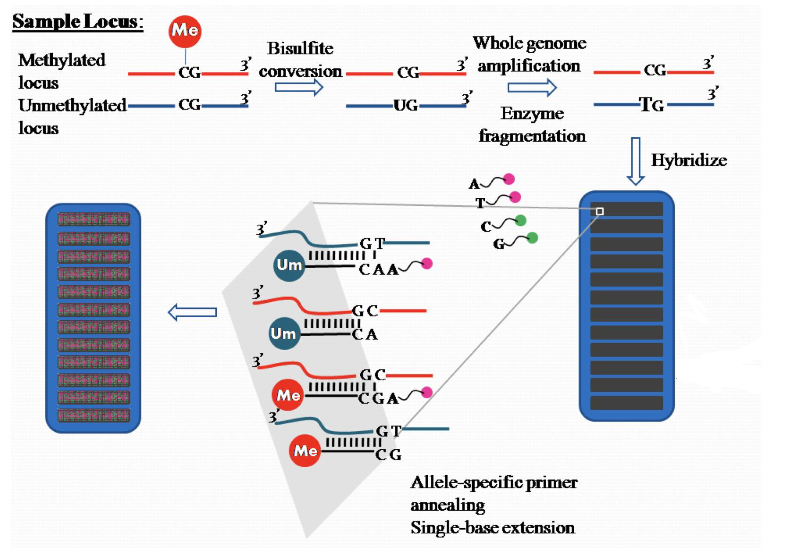
850K芯片采用了两种探针Infinium Ⅰ 和Infinium Ⅱ对样品甲基化进行测定,Infinium I采用了两种bead(甲基化M和非甲基化U,如图显示),而II只有一种bead(即甲基化和非甲基化在一起),这也导致了它们在后续荧光探测的不同,而根据不同探针的bead的荧光值,就可以得到样品各个位点上的甲基化水平。
|
|
|
850K芯片采用了两种探针Infinium Ⅰ 和Infinium Ⅱ对样品甲基化进行测定,Infinium I采用了两种bead(甲基化M和非甲基化U,如图显示),而II只有一种bead(即甲基化和非甲基化在一起),这也导致了它们在后续荧光探测的不同,而根据不同探针的bead的荧光值,就可以得到样品各个位点上的甲基化水平。
|
|
|
|
|
|
|
|
|
###APP简介
|
|
|
|
|
|
|
|
|
#### 
|
|
|
|
|
|
|
|
|
|
|
|
对于位点甲基化水平的定量测量通常使用两种指标:β值和M值
|
|
|
|
|
|
|
|
|
|
|
|
β值:
|
|
|
|
|
|
|
|
|
|
|
|
(其中M代表甲基化信号,U代表非甲基化信号)
|
|
|
|
|
|
|
|
|
|
|
|
M值:
|
|
|
|
|
|
|
|
|
|
|
|
β值是最常用的甲基化水平的定量方式,可以直观地了解不同位点的甲基化水平;而M值具有更好的统计学特性,更适用于对样品数据的统计分析。
|
|
|
|
|
|
|
|
|
|
|
|
#### APP简介
|
|
|
|
|
|
|
|
|
为了更好更便捷的分析全基因组甲基化数据,我们选用了分析850K芯片的R包——minfi包,构建了分析pipeline,可以得到全基因组的各个位点甲基化表达谱。
|
|
|
为了更好更便捷的分析全基因组甲基化数据,我们选用了分析850K芯片的R包——minfi包,构建了分析pipeline,可以得到全基因组的各个位点甲基化表达谱。
|
|
|
|
|
|
|
|
|
#流程和参数
|
|
|
|
|
|
|
|
|
### 流程和参数
|
|
|
|
|
|
|
|
|
#### 850K array分析流程
|
|
|
#### 850K array分析流程
|
|
|
|
|
|
|
|
|
#输入和输出
|
|
|
|
|
|
|
|
|

|
|
|
|
|
|
|
|
|
###输入
|
|
|
|
|
|
|
|
|
本APP将EPIC芯片的原始数据读入后,过滤了p<0.05的列和p<0.01的行后使用Funnorm方法对数据进行归一化,再过滤其中的SNP位点和性染色体位点,最终得到四张表格(详见输出部分)
|
|
|
|
|
|
|
|
|
|
|
|
### 使用方法
|
|
|
|
|
|
|
|
|
|
|
|
按照上述步骤安装成功之后,就可以通过下面的命令使用APP
|
|
|
|
|
|
|
|
|
|
|
|
`$ Rscript EPIC.modified.R -p xxx -i xxx `
|
|
|
|
|
|
|
|
|
|
|
|
具体参数要求见输入模块
|
|
|
|
|
|
|
|
|
|
|
|
### 输入和输出
|
|
|
|
|
|
|
|
|
|
|
|
#### 输入
|
|
|
|
|
|
|
|
|
需要一个文件夹,其中包含:
|
|
|
需要一个文件夹,其中包含:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
▪ sample_sheet文件:样本的注释信息文件,命名为“sample_sheet.csv”,其中包含Sample_Name,Sentrix_ID, Sentrix_Position, Sample_Group等注释信息
|
|
|
▪ sample_sheet文件:样本的注释信息文件,命名为“sample_sheet.csv”,其中包含Sample_Name,Sentrix_ID, Sentrix_Position, Sample_Group等注释信息
|
|
|
|
|
|
|
|
|
###输出
|
|
|
|
|
|
|
|
|
#### 输出
|
|
|
|
|
|
|
|
|
|
|
|
任务完成结束后,便可以在阿里云相应的OSS端生成相应的文件,该输出包含四张含有所有样本M值和beta值的表格(行名为各位点标号,列名为各样本名)。
|
|
|
|
|
|
|
|
|
|
|
|
▪ raw.mVal.txt(原始数据(文件读入后直接得到)的M值)
|
|
|
|
|
|
|
|
|
|
|
|
▪ raw.bVal.txt(原始数据的β值)
|
|
|
|
|
|
|
|
|
|
|
|
▪ filter.p.sex.snp.mVal.txt(过滤和归一化(过滤了低质量位点、SNP位点和性染色体位点,使用Funnorm进行归一化)后数据的M值)
|
|
|
|
|
|
|
|
|
在850K芯片的分析中,beta 值是最常用的甲基化水平的定量方式,其主要用于差异分析。
|
|
|
|
|
|
|
|
|
**▪** filter.p.sex.snp.bVal.txt(过滤和归一化后数据的β值)
|
|
|
|
|
|
|

 LUYAO REN
5 years ago
LUYAO REN
5 years ago
{kind=link}
{kind=link}
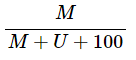
{kind=link}
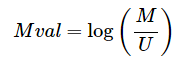
{kind=link}
