
 LUYAO REN
212960e46d
region
LUYAO REN
212960e46d
region
|
4 yıl önce | |
|---|---|---|
| picture | 4 yıl önce | |
| tasks | 4 yıl önce | |
| README.md | 4 yıl önce | |
| defaults | 4 yıl önce | |
| inputs | 4 yıl önce | |
| workflow.wdl | 4 yıl önce | |
README.md
DeepVariant
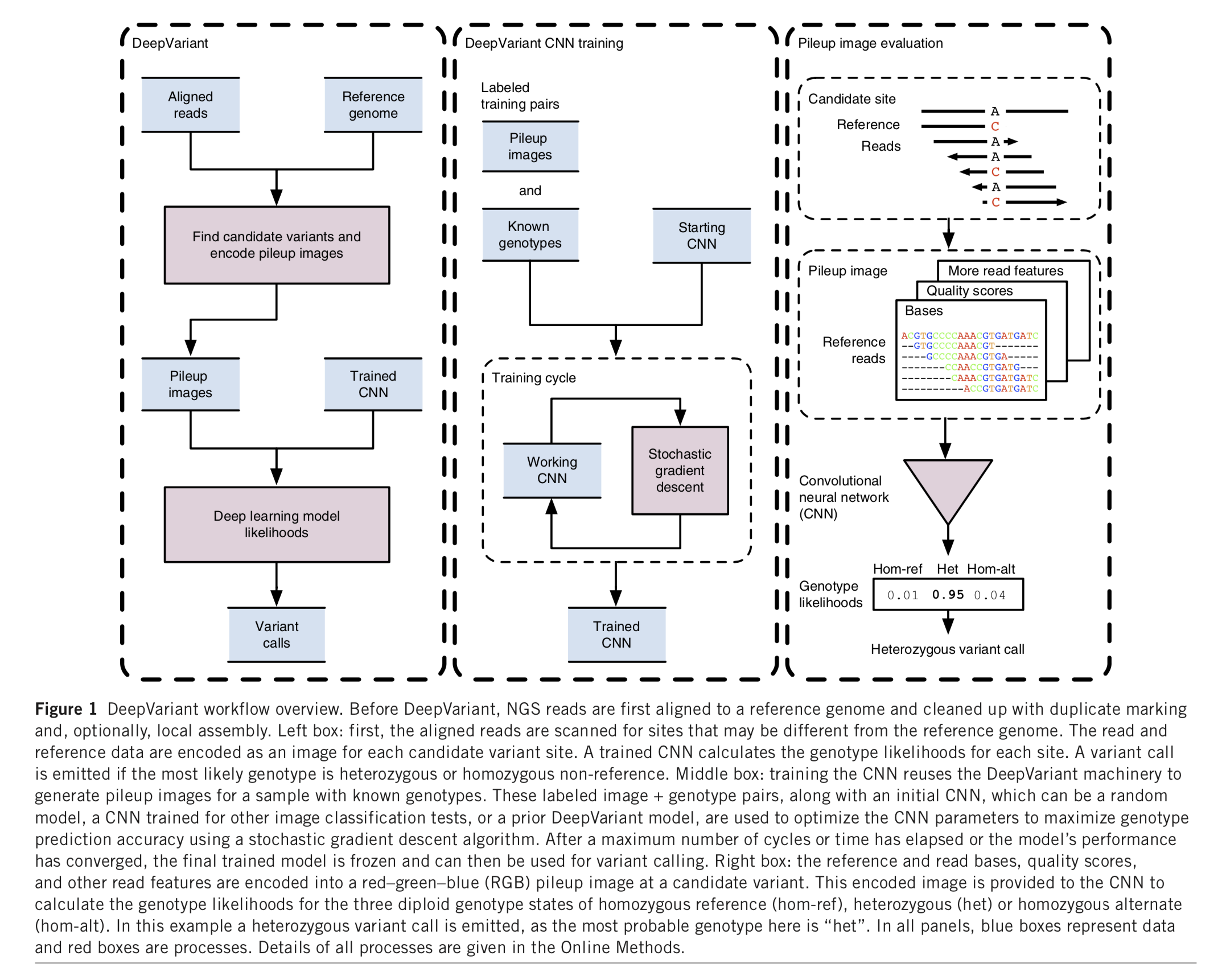
DeepVariant was trained on 8 whole genome replicates of NA12878 sequenced under a variety of conditions related to library preparation. These conditions include loading concentration, library size selection, and laboratory technician. Then the trained model can be generalized to a variety of new datasets and call variants.
Docker uploaded to Alibaba Cloud is downloaded from dockerhub (version r0.8.0).
WGS training datasets v0.8 include 12 HG001 PCR-free, 2 HG005 PCR-free, 4 HG001 PCR+. Sequencing platforms are all Illumina Hiseq. For Illumina Novaseq, BGISEQ-500 and BGISEQ-2000, we probably need to train customized small variants callers, more information can be found in Github.
[CUSTOMIZED MODELS TO BE ADDED]
DeepVariant pipeline consist of 3 steps:
make_examplesconsumes reads and the refernece genome to create TensorFlow examples for evaluation with deep learning models.call_variants(Multiple-threads) consums TFRecord files of tf.Examples protos created bymake_examplesand a deep learning model checkpoint and evaluates the model on each example in the TFRecord. The output here is a TFRecord of CallVariantOutput protos. Multiple-threadspostprocess_variants(Single-thread) reads all of the output TFRecord files fromcall_variants, it needs to see all of the outputs fromcall_variantsfor a single sample to merge into a final VCF.
Some tips for DeepVariant:
- Duplicate marking may be performed, there is almost no difference inaccuracy except lower (<20x) coverages.
- Authors recommend that you do not perform BQSR. Running BQSR has a small decrease on accuracy.
- It is not necessary to do any form of indel realignment, though there is not a difference in DeepVariant accuracy either way.
You can run with one common using the run_deepvariant.py script
python run_deepvariant.py --model_type=WGS \
--ref=../../data/"${REFERENCE_FILE}" \
--reads=../../data/"${BAM_FILE}" \
--regions "chr20:10,000,000-10,010,000" \
--output_vcf=../output/output.vcf.gz \
--output_gvcf=../output/output.g.vcf.gz
Four files are generated
output.vcf.gz
output.vcf.gz.tbi
output.g.vcf.gz
output.g.vcf.gz.tbi
Command used in choppy app
python run_deepvariant.py --model_type=WGS \
--ref=${ref_dir}/${fasta} \
--reads=${Dedup.bam} \
--output_vcf=${sample}_DP.vcf.gz
Reference:
DeepVariant Github https://github.com/google/deepvariant
DeepVariant paper https://www.nature.com/articles/nbt.4235