
共有 9 个文件被更改,包括 73 次插入 和 26 次删除
+ 7
- 19
README.md
查看文件
| ### 甲基化原理简述 | ### 甲基化原理简述 | ||||
|  | |||||
| 全基因组亚硫酸氢盐测序用于研究基因粒度的DNA甲基化模式。 亚硫酸氢盐处理将胞嘧啶转化为尿嘧啶,但甲基化胞嘧啶不变。 | 全基因组亚硫酸氢盐测序用于研究基因粒度的DNA甲基化模式。 亚硫酸氢盐处理将胞嘧啶转化为尿嘧啶,但甲基化胞嘧啶不变。 | ||||
| 比对软件(如Bismark)将基因组序列转化之后进行比对。 | 比对软件(如Bismark)将基因组序列转化之后进行比对。 | ||||
|  | |||||
| ### APP功能简述 | ### APP功能简述 | ||||
| 为了更好的分析全基因组甲基化数据,我们选用了目前最好的比对软件Bismark,构建了分析pipeline。用来提取全基因组的CpG,CHH,CHG甲基化模式信息。 | 为了更好的分析全基因组甲基化数据,我们选用了目前最好的比对软件Bismark,构建了分析pipeline。用来提取全基因组的CpG,CHH,CHG甲基化模式信息。 | ||||
| ### WGBS分析流程 | ### WGBS分析流程 | ||||
|  | |||||
|  | |||||
| ### 参数说明 | ### 参数说明 | ||||
| -p 多线程 | -p 多线程 | ||||
| ``` | |||||
| ## 输入和输出 | ## 输入和输出 | ||||
| 以CpG_context_test_data_bismark_bt2.txt为例 | 以CpG_context_test_data_bismark_bt2.txt为例 | ||||
| ``` | ``` | ||||
| Bismark methylation extractor version v0.19.0 | Bismark methylation extractor version v0.19.0 | ||||
| SRR15024317_length=86 - 1 57798691 z | SRR15024317_length=86 - 1 57798691 z | ||||
| SRR15024319_length=86 + 2 10166600 Z | SRR15024319_length=86 + 2 10166600 Z | ||||
| SRR15024331_length=86 + 11 77736289 Z | SRR15024331_length=86 + 11 77736289 Z | ||||
| SRR15024338_length=86 + 3 197272186 Z | SRR15024338_length=86 + 3 197272186 Z | ||||
| ``` | ``` | ||||
| 第一行为Bismark的版本信息 | 第一行为Bismark的版本信息 | ||||
| 不同字母表明不同的甲基化状态: | 不同字母表明不同的甲基化状态: | ||||
| ``` | ``` | ||||
| X 代表CHG中甲基化的C | X 代表CHG中甲基化的C | ||||
| x 代笔CHG中非甲基化的C | x 代笔CHG中非甲基化的C | ||||
| H 代表CHH中甲基化的C | H 代表CHH中甲基化的C | ||||
| z 代表CpG中非甲基化的C | z 代表CpG中非甲基化的C | ||||
| U 代表其他情况的甲基化C(CN或者CHN) | U 代表其他情况的甲基化C(CN或者CHN) | ||||
| u 代表其他情况的非甲基化C (CN或者CHN) | u 代表其他情况的非甲基化C (CN或者CHN) | ||||
| ``` | ``` | ||||
| #### 补充文件 | #### 补充文件 | ||||
| 上面的文件是methylation calling 最直接的证据,但是对于甲基化水平的定量来说,缺少了相关信息。运行bismark_methylation_extractor时,除了生成上述文件之外,还会有下列3个文件: | 上面的文件是methylation calling 最直接的证据,但是对于甲基化水平的定量来说,缺少了相关信息。运行bismark_methylation_extractor时,除了生成上述文件之外,还会有下列3个文件: | ||||
| ``` | |||||
| test_data_bismark_bt2_splitting_report.txt | test_data_bismark_bt2_splitting_report.txt | ||||
| test_data_bismark_bt2.M-bias.txt | test_data_bismark_bt2.M-bias.txt | ||||
| test_data_bismark_bt2.M-bias_R1.png | test_data_bismark_bt2.M-bias_R1.png | ||||
| ``` | |||||
| ##### test_data_bismark_bt2_splitting_report.txt | ##### test_data_bismark_bt2_splitting_report.txt | ||||
| 记录了该样本甲基化的汇总信息 | 记录了该样本甲基化的汇总信息 | ||||
| ``` | ``` | ||||
| Final Cytosine Methylation Report | Final Cytosine Methylation Report | ||||
| Total number of C’s analysed: 40348 | Total number of C’s analysed: 40348 | ||||
| Total methylated C’s in CpG context: 1365 | Total methylated C’s in CpG context: 1365 | ||||
| C methylated in CpG context: 66.8% | C methylated in CpG context: 66.8% | ||||
| C methylated in CHG context: 0.2% | C methylated in CHG context: 0.2% | ||||
| C methylated in CHH context: 0.4% | C methylated in CHH context: 0.4% | ||||
| ``` | ``` | ||||
| ##### test_data_bismark_bt2.M-bias.txt | ##### test_data_bismark_bt2.M-bias.txt | ||||
| 部分文件内容如下 | 部分文件内容如下 | ||||
| ``` | ``` | ||||
| CpG context | CpG context | ||||
| position count methylated count unmethylated % methylation coverage | position count methylated count unmethylated % methylation coverage | ||||
| 1 42 13 76.36 55 | 1 42 13 76.36 55 | ||||
| 2 31 9 77.50 40 | 2 31 9 77.50 40 | ||||
| ``` | ``` | ||||
+ 2
- 4
inputs
查看文件
| { | { | ||||
| "{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", | |||||
| "{{ project_name }}.ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", | |||||
| "{{ project_name }}.ref_dir": "oss://pgx-test-data/wgbs/genome/hg38/", | |||||
| "{{ project_name }}.fastq_1": "{{ read1 }}", | "{{ project_name }}.fastq_1": "{{ read1 }}", | ||||
| "{{ project_name }}.cluster_config": "{{ cluster if cluster != '' else 'OnDemand ecs.sn2ne.2xlarge img-ubuntu-vpc' }}", | "{{ project_name }}.cluster_config": "{{ cluster if cluster != '' else 'OnDemand ecs.sn2ne.2xlarge img-ubuntu-vpc' }}", | ||||
| "{{ project_name }}.docker": "registry.cn-shanghai.aliyuncs.com/pgx-docker-registry/sentieon-genomics:v2018.08.01", | |||||
| "{{ project_name }}.docker": "registry-vpc.cn-shanghai.aliyuncs.com/pgx-docker-registry/bismark", | |||||
| "{{ project_name }}.sample": "{{ sample_name }}", | "{{ project_name }}.sample": "{{ sample_name }}", | ||||
| "{{ project_name }}.disk_size": "{{ disk_size }}", | "{{ project_name }}.disk_size": "{{ disk_size }}", | ||||
| "{{ project_name }}.regions": "{{ regions }}", | |||||
| "{{ project_name }}.fastq_2": "{{ read2 }}" | "{{ project_name }}.fastq_2": "{{ read2 }}" | ||||
| } | } | ||||
二进制
picture/BS原理.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|

|
|
| 宽度: 964 | 高度: 485 | 大小: 33KB |
二进制
picture/Thumbs.db
查看文件
二进制
picture/WGBS流程.png
查看文件
{kind=link}
| 之前 | 之后 |
|---|---|
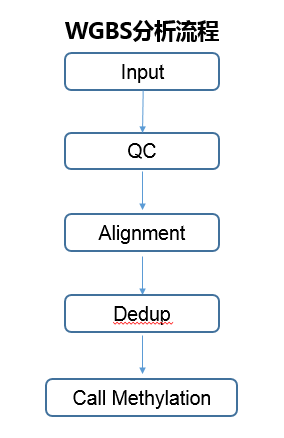
|
|
| 宽度: 284 | 高度: 438 | 大小: 8.8KB |
+ 0
- 1
tasks/Dedup.wdl
查看文件
| command <<< | command <<< | ||||
| set -o pipefail | set -o pipefail | ||||
| set -e | set -e | ||||
| nt=$(nproc) | |||||
| deduplicate_bismark -p --bam ${unsorted_bam} | deduplicate_bismark -p --bam ${unsorted_bam} | ||||
| >>> | >>> | ||||
| runtime { | runtime { |
+ 34
- 0
tasks/Dedup.wdl.bak
查看文件
| task Dedup { | |||||
| String sample | |||||
| File unsorted_bam | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| deduplicate_bismark -p --bam ${unsorted_bam} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File Dedup_bam = "${sample}_R1_val_1_bismark_bt2_pe.deduplicated.bam" | |||||
| } | |||||
| } | |||||
+ 1
- 2
tasks/Mapping.wdl
查看文件
| command <<< | command <<< | ||||
| set -o pipefail | set -o pipefail | ||||
| set -e | set -e | ||||
| nt=$(nproc) | |||||
| bismark --bowtie2 -p ${nt} --bam ${ref_dir} -1 ${trim_read1} -2 ${trim_read2} | |||||
| bismark --bowtie2 -p 8 --bam ${ref_dir} -1 ${trim_read1} -2 ${trim_read2} | |||||
| >>> | >>> | ||||
+ 29
- 0
tasks/Mapping.wdl.bak
查看文件
| task mapping { | |||||
| File ref_dir | |||||
| File trim_read1 | |||||
| File trim_read2 | |||||
| String sample | |||||
| String docker | |||||
| String cluster_config | |||||
| String disk_size | |||||
| command <<< | |||||
| set -o pipefail | |||||
| set -e | |||||
| nt=$(nproc) | |||||
| bismark --bowtie2 -p 8 --bam ${ref_dir} -1 ${trim_read1} -2 ${trim_read2} | |||||
| >>> | |||||
| runtime { | |||||
| docker:docker | |||||
| cluster: cluster_config | |||||
| systemDisk: "cloud_ssd 40" | |||||
| dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" | |||||
| } | |||||
| output { | |||||
| File unsorted_bam = "${sample}_R1_val_1_bismark_bt2_pe.bam" | |||||
| } | |||||
| } |
正在加载...