|
|
|
@@ -0,0 +1,179 @@ |
|
|
|
## README |
|
|
|
|
|
|
|
**Author:** Huang Yechao |
|
|
|
|
|
|
|
**E-mail:**17210700095@fudan.edu.cn |
|
|
|
|
|
|
|
**Git:** http://choppy.3steps.cn/huangyechao/target-germline.git |
|
|
|
|
|
|
|
**Last Updates:** 16/1/2019 |
|
|
|
|
|
|
|
**Description** |
|
|
|
|
|
|
|
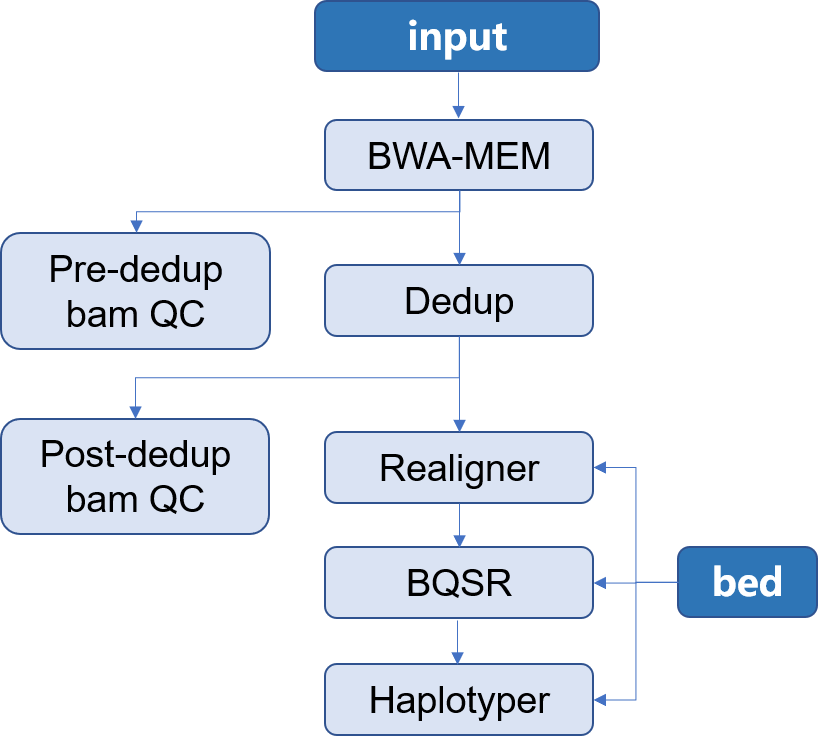
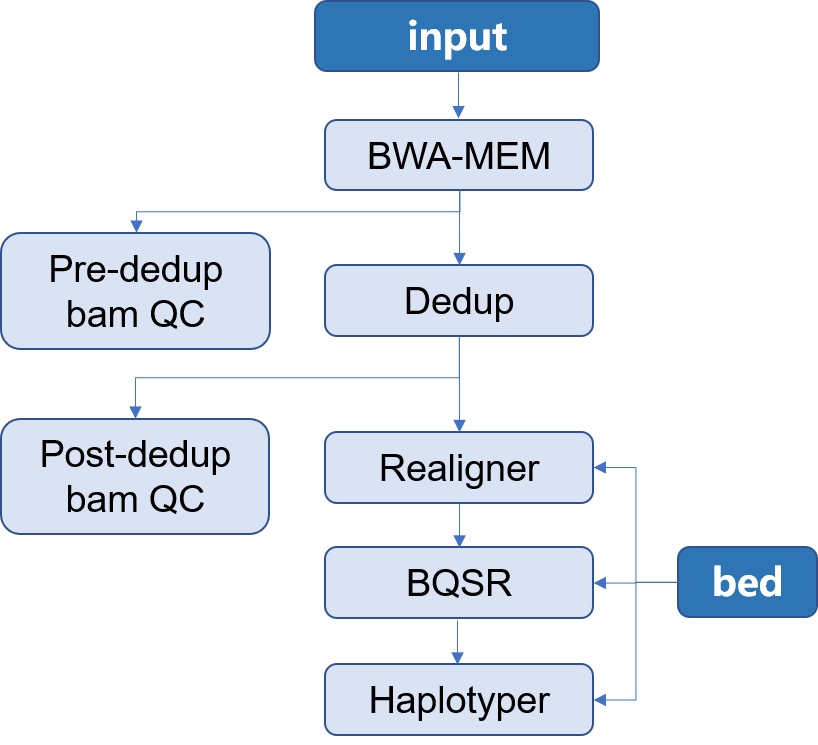
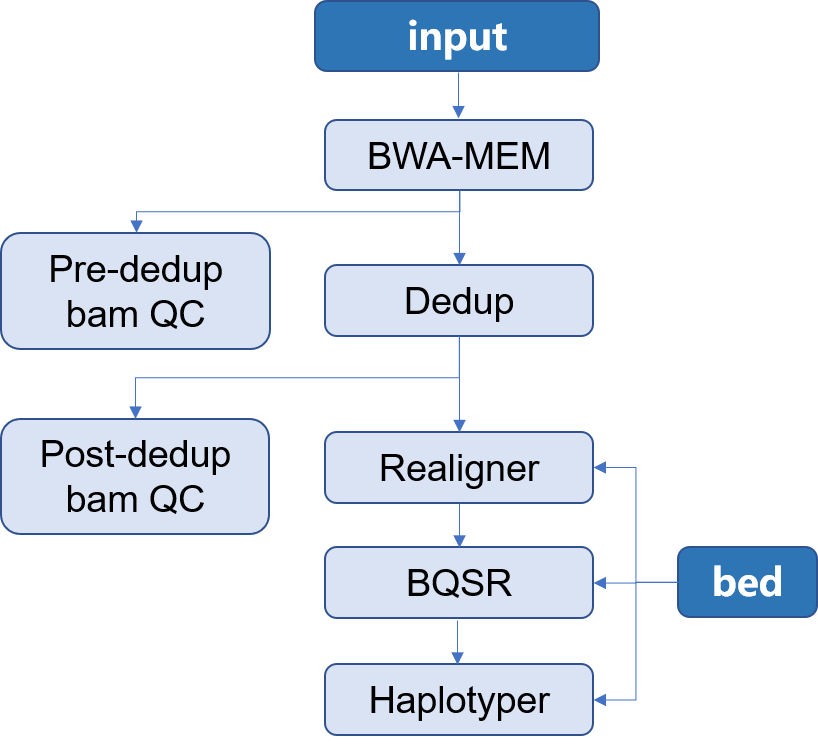
> 本 APP 所构建的是用于二代测序目标区域测序 Germline 分析流程。使用的软件是[Sentieon](http://goldenhelix.com/products/sentieon/index.html):*A fast and accurate solution to variant calling from next-generation sequence data* 。本流程构建所使用的方法是基于流程语言WDL 并将其封装为[Choppy](http://docs.3steps.cn)平台上的APP进行使用。流程图如下所示: |
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
|
1. input:通常为二代目标区域测序所获得的fastq文件,通常包含两个数据文件`R1`和`R2`;此外还应当包含有测序时使用的 `bed` 文件 |
|
|
|
|
|
|
|
2. Mapping:将测序所得的数据与参考基因组进行比对,找到每一条read在参考基因组上的位置,将结果信息储存在**bam**文件中,并对获得的 **bam** 文件进行质控 |
|
|
|
3. Dedup:在制备文库的过程中,由于PCR扩增过程中会存在一些偏差,有的序列会被过量扩增。在比对的时候,这些过量扩增出来的完全相同的序列就会比对到基因组的相同位置。而这些过量扩增的reads并不是基因组自身固有序列,不能作为变异检测的证据,因此,要尽量去除这些由PCR扩增所形成的duplicates,并对去除重复之后的**bam**文件进行质控 |
|
|
|
4. Realigner: 将比对到 indel 附近的 reads 进行局部重新比对,将比对的错误率降到最低 |
|
|
|
5. BQSR:对bam文件里reads的碱基质量值进行重新校正,使最后输出的bam文件中reads中碱基的质量值能够更加接近真实的与参考基因组之间错配的概率 |
|
|
|
6. Haplotyer:变异检测,主要包括 **SNP** 和 **INDEL** |
|
|
|
|
|
|
|
### APP 内容简介 |
|
|
|
|
|
|
|
1. `tasks`目录中分析流程中每一个步骤的 **WDL** 文件,如 `mapping.wdl` 如下所示 |
|
|
|
|
|
|
|
```bash |
|
|
|
task mapping { |
|
|
|
String fasta |
|
|
|
File ref_dir |
|
|
|
File fastq_1 |
|
|
|
File fastq_2 |
|
|
|
|
|
|
|
String SENTIEON_INSTALL_DIR |
|
|
|
String group |
|
|
|
String sample |
|
|
|
String pl |
|
|
|
String docker |
|
|
|
String cluster_config |
|
|
|
String disk_size |
|
|
|
|
|
|
|
command <<< |
|
|
|
set -o pipefail |
|
|
|
set -e |
|
|
|
export SENTIEON_LICENSE=192.168.0.55:8990 |
|
|
|
nt=$(nproc) |
|
|
|
${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i - |
|
|
|
>>> |
|
|
|
|
|
|
|
runtime { |
|
|
|
dockerTag:docker |
|
|
|
cluster: cluster_config |
|
|
|
systemDisk: "cloud_ssd 40" |
|
|
|
dataDisk: "cloud_ssd " + disk_size + " /cromwell_root/" |
|
|
|
} |
|
|
|
output { |
|
|
|
File sorted_bam = "${sample}.sorted.bam" |
|
|
|
File sorted_bam_index = "${sample}.sorted.bam.bai" |
|
|
|
} |
|
|
|
} |
|
|
|
|
|
|
|
``` |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2. `workflow.wdl` 是定义了每一个步骤的输入文件以及各个步骤之间的以来关系的文件: |
|
|
|
|
|
|
|
```bash |
|
|
|
import "./tasks/mapping.wdl" as mapping |
|
|
|
import "./tasks/Metrics.wdl" as Metrics |
|
|
|
import "./tasks/Dedup.wdl" as Dedup |
|
|
|
import "./tasks/deduped_Metrics.wdl" as deduped_Metrics |
|
|
|
import "./tasks/Realigner.wdl" as Realigner |
|
|
|
import "./tasks/BQSR.wdl" as BQSR |
|
|
|
import "./tasks/Haplotyper.wdl" as Haplotyper |
|
|
|
|
|
|
|
|
|
|
|
workflow {{ project_name }} { |
|
|
|
|
|
|
|
File fastq_1 |
|
|
|
File fastq_2 |
|
|
|
|
|
|
|
String SENTIEON_INSTALL_DIR |
|
|
|
String sample |
|
|
|
String docker |
|
|
|
|
|
|
|
String fasta |
|
|
|
File ref_dir |
|
|
|
File dbmills_dir |
|
|
|
String db_mills |
|
|
|
File dbsnp_dir |
|
|
|
File region |
|
|
|
String dbsnp |
|
|
|
String disk_size |
|
|
|
String cluster_config |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
call mapping.mapping as mapping { |
|
|
|
input: |
|
|
|
SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, |
|
|
|
group=sample, |
|
|
|
sample=sample, |
|
|
|
pl="ILLUMINAL", |
|
|
|
fasta=fasta, |
|
|
|
ref_dir=ref_dir, |
|
|
|
fastq_1=fastq_1, |
|
|
|
fastq_2=fastq_2, |
|
|
|
docker=docker, |
|
|
|
disk_size=disk_size, |
|
|
|
cluster_config=cluster_config |
|
|
|
} |
|
|
|
|
|
|
|
call Metrics.Metrics as Metrics { |
|
|
|
input: |
|
|
|
SENTIEON_INSTALL_DIR=SENTIEON_INSTALL_DIR, |
|
|
|
fasta=fasta, |
|
|
|
ref_dir=ref_dir, |
|
|
|
sorted_bam=mapping.sorted_bam, |
|
|
|
sorted_bam_index=mapping.sorted_bam_index, |
|
|
|
sample=sample, |
|
|
|
docker=docker, |
|
|
|
disk_size=disk_size, |
|
|
|
cluster_config=cluster_config |
|
|
|
} |
|
|
|
...... |
|
|
|
...... |
|
|
|
} |
|
|
|
|
|
|
|
``` |
|
|
|
|
|
|
|
其中文件最上面的 `import` 代表了所要使用的task文件,中间部分`File/String xxx` 表明了任务所传递出需要定义变量及其类型,`call`部分声明了流程的各个步骤及其依赖关系。(文档的具体说明详见[WDL](https://software.broadinstitute.org/wdl/documentation/spec#alternative-heredoc-syntax)) |
|
|
|
|
|
|
|
3. `input` 文件为整个 **APP** 运行时所要输入的参数,对于可以固定的参数可以直接在`input`文件中给出,对于需要改变的参数用`{{}}`进行引用,将会使得参数在 `samples` 文件中出现;其中`project_name`为所运行的任务的名称,需要在提交任务是进行定义 |
|
|
|
|
|
|
|
```bash |
|
|
|
{ |
|
|
|
"{{ project_name }}.fasta": "GRCh38.d1.vd1.fa", |
|
|
|
"{{ project_name }}.ref_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", |
|
|
|
"{{ project_name }}.dbsnp": "dbsnp_146.hg38.vcf", |
|
|
|
"{{ project_name }}.fastq_1": "{{ read1 }}", |
|
|
|
"{{ project_name }}.SENTIEON_INSTALL_DIR": "/opt/sentieon-genomics", |
|
|
|
"{{ project_name }}.dbmills_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", |
|
|
|
"{{ project_name }}.db_mills": "Mills_and_1000G_gold_standard.indels.hg38.vcf", |
|
|
|
"{{ project_name }}.cluster_config": "{{ cluster if cluster != '' else 'OnDemand ecs.sn2ne.2xlarge img-ubuntu-vpc' }}", |
|
|
|
"{{ project_name }}.docker": "localhost:5000/sentieon-genomics:v2018.08.01 oss://pgx-docker-images/dockers", |
|
|
|
"{{ project_name }}.dbsnp_dir": "oss://pgx-reference-data/GRCh38.d1.vd1/", |
|
|
|
"{{ project_name }}.sample": "{{ sample_name }}", |
|
|
|
"{{ project_name }}.disk_size": "{{ disk_size }}", |
|
|
|
"{{ project_name }}.regions": "{{ regions }}", |
|
|
|
"{{ project_name }}.fastq_2": "{{ read2 }}" |
|
|
|
} |
|
|
|
``` |
|
|
|
|
|
|
|
> `{{ cluster if cluster != '' else 'OnDemand ecs.sn2ne.2xlarge img-ubuntu-vpc' }}`表示当没有指定`cluster` 的配置信息时,则默认使用 **ecs.sn2ne.2xlarge** |
|
|
|
|
|
|
|
4. `sample.csv` 文件为提交任务时使用的输入文件,其内容是根据`input`文件中定义的信息对应生成的,也可使用 `Choppy` 的 `samples` 功能生成: |
|
|
|
|
|
|
|
```bash |
|
|
|
choppy samples target-germline --output samples.csv |
|
|
|
``` |
|
|
|
|
|
|
|
```bash |
|
|
|
#### samples.csv |
|
|
|
read1,read2,regions,sample_name,cluster,disk_size,sample_id |
|
|
|
``` |
|
|
|
|
|
|
|
其中`sample_id`对应于所分析样本的索引号,用于生成当前样本提交时的任务信息,应注意不要包含`_`,否则会出现报错。 |
|
|
|
|
|
|
|
5. 提交任务 |
|
|
|
|
|
|
|
```bash |
|
|
|
choppy batch target-germline samples.csv --project-name your_project |
|
|
|
``` |
|
|
|
|
|
|
|
(更多使用信息,详见[Choppy使用说明](http://docs.3steps.cn)) |
|
|
|
|


{kind=link}
{kind=link}
{kind=link}